

Meraculous: de novo genome assembly with short paired-end reads
- PMID: 21876754
- PMCID: PMC3158087
- DOI: 10.1371/journal.pone.0023501
Meraculous: de novo genome assembly with short paired-end reads
Abstract
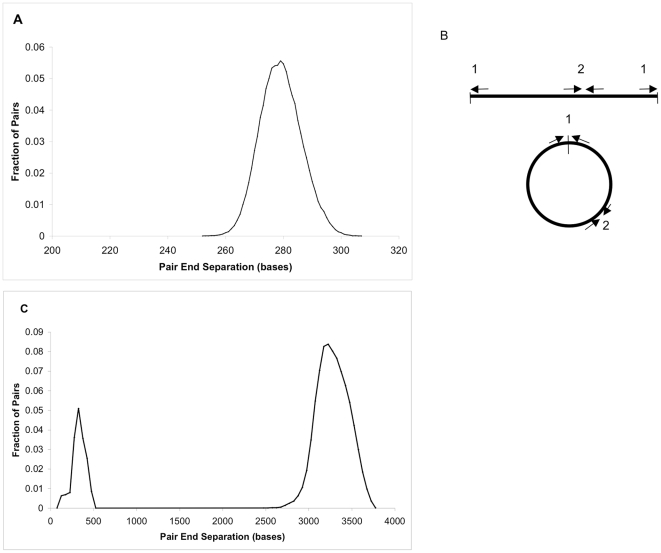
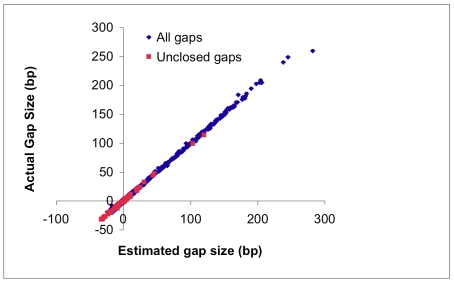
We describe a new algorithm, meraculous, for whole genome assembly of deep paired-end short reads, and apply it to the assembly of a dataset of paired 75-bp Illumina reads derived from the 15.4 megabase genome of the haploid yeast Pichia stipitis. More than 95% of the genome is recovered, with no errors; half the assembled sequence is in contigs longer than 101 kilobases and in scaffolds longer than 269 kilobases. Incorporating fosmid ends recovers entire chromosomes. Meraculous relies on an efficient and conservative traversal of the subgraph of the k-mer (deBruijn) graph of oligonucleotides with unique high quality extensions in the dataset, avoiding an explicit error correction step as used in other short-read assemblers. A novel memory-efficient hashing scheme is introduced. The resulting contigs are ordered and oriented using paired reads separated by ∼280 bp or ∼3.2 kbp, and many gaps between contigs can be closed using paired-end placements. Practical issues with the dataset are described, and prospects for assembling larger genomes are discussed.
Conflict of interest statement
Figures
References
-
- Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet. 2010;11:31–46. - PubMed
-
- Bentley DR. Whole-genome re-sequencing. Curr Opin Genet Dev. 2006;16:545–552. - PubMed
-
- Wheeler DA, Srinivasan M, Egholm M, Shen Y, Chen L, et al. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008;452:872–876. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
