

Two new Rett syndrome families and review of the literature: expanding the knowledge of MECP2 frameshift mutations
- PMID: 21878110
- PMCID: PMC3173288
- DOI: 10.1186/1750-1172-6-58
Two new Rett syndrome families and review of the literature: expanding the knowledge of MECP2 frameshift mutations
Abstract
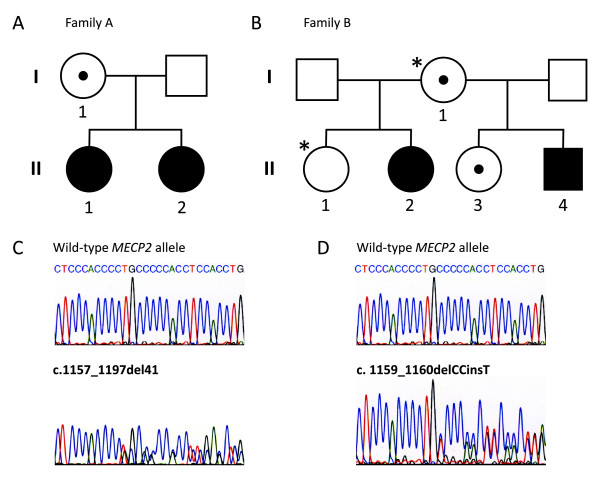
Background: Rett syndrome (RTT) is an X-linked dominant neurodevelopmental disorder, which is usually caused by de novo mutations in the MECP2 gene. More than 70% of the disease causing MECP2 mutations are eight recurrent C to T transitions, which almost exclusively arise on the paternally derived X chromosome. About 10% of the RTT cases have a C-terminal frameshift deletion in MECP2. Only few RTT families with a segregating MECP2 mutation, which affects female carriers with a phenotype of mental retardation or RTT, have been reported in the literature. In this study we describe two new RTT families with three and four individuals, respectively, and review the literature comparing the type of mutations and phenotypes observed in RTT families with those observed in sporadic cases. Based on these observations we also investigated origin of mutation segregation to further improve genetic counselling.
Methods: MECP2 mutations were identified by direct sequencing. XCI studies were performed using the X-linked androgen receptor (AR) locus. The parental origin of de novo MECP2 frameshift mutations was investigated using intronic SNPs.
Results: In both families a C-terminal frameshift mutation segregates. Clinical features of the mutation carriers vary from classical RTT to mild mental retardation. XCI profiles of the female carriers correlate to their respective geno-/phenotypes. The majority of the de novo frameshift mutations occur on the paternally derived X chromosome (7/9 cases), without a paternal age effect.
Conclusions: The present study suggests a correlation between the intrafamilial phenotypic differences observed in RTT families and their respective XCI pattern in blood, in contrast to sporadic RTT cases where a similar correlation has not been demonstrated. Furthermore, we found de novo MECP2 frameshift mutations frequently to be of paternal origin, although not with the same high paternal occurrence as in sporadic cases with C to T transitions. This suggests further investigations of more families. This study emphasizes the need for thorough genetic counselling of families with a newly diagnosed RTT patient.
Figures
Similar articles
-
[Analysis of the parental origin of de novo MECP2 mutations and X chromosome inactivation in fifteen sporadic cases with Rett syndrome].Zhonghua Er Ke Za Zhi. 2009 Aug;47(8):565-9. Zhonghua Er Ke Za Zhi. 2009. PMID: 19951486 Chinese.
-
[X chromosome inactivation patterns in patients with Rett syndrome and their mothers and the parental origin of the priority inactive X chromosome].Zhonghua Er Ke Za Zhi. 2006 Sep;44(9):648-52. Zhonghua Er Ke Za Zhi. 2006. PMID: 17217653 Chinese.
-
[Genetic features and mechanism of Rett syndrome in Chinese population].Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2014 Feb;31(1):1-5. doi: 10.3760/cma.j.issn.1003-9406.2014.01.001. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2014. PMID: 24510551 Chinese.
-
Rett syndrome: the complex nature of a monogenic disease.J Mol Med (Berl). 2003 Jun;81(6):346-54. doi: 10.1007/s00109-003-0444-9. Epub 2003 May 16. J Mol Med (Berl). 2003. PMID: 12750821 Review.
-
Rett syndrome: from the gene to the disease.Eur Neurol. 2009;61(1):3-10. doi: 10.1159/000165342. Epub 2008 Oct 24. Eur Neurol. 2009. PMID: 18948693 Review.
Cited by
-
Suppressor mutations in Mecp2-null mice implicate the DNA damage response in Rett syndrome pathology.Genome Res. 2020 Apr;30(4):540-552. doi: 10.1101/gr.258400.119. Epub 2020 Apr 21. Genome Res. 2020. PMID: 32317254 Free PMC article.
-
Genetics, molecular biology, and phenotypes of x-linked epilepsy.Mol Neurobiol. 2014 Jun;49(3):1166-80. doi: 10.1007/s12035-013-8589-1. Epub 2013 Nov 22. Mol Neurobiol. 2014. PMID: 24258407 Review.
-
MECP2 Duplication Syndrome: Evidence of Enhanced Oxidative Stress. A Comparison with Rett Syndrome.PLoS One. 2016 Mar 1;11(3):e0150101. doi: 10.1371/journal.pone.0150101. eCollection 2016. PLoS One. 2016. PMID: 26930212 Free PMC article.
-
Normalized Clinical Severity Scores Reveal a Correlation between X Chromosome Inactivation and Disease Severity in Rett Syndrome.Genes (Basel). 2024 May 8;15(5):594. doi: 10.3390/genes15050594. Genes (Basel). 2024. PMID: 38790223 Free PMC article.
-
Pluripotent stem cells as a model to study non-coding RNAs function in human neurogenesis.Front Cell Neurosci. 2013 Aug 27;7:140. doi: 10.3389/fncel.2013.00140. eCollection 2013. Front Cell Neurosci. 2013. PMID: 23986659 Free PMC article.
References
-
- Hagberg B, Hanefeld F, Percy A, Skjeldal O. An update on clinically applicable diagnostic criteria in Rett syndrome. Comments to Rett Syndrome Clinical Criteria Consensus Panel Satellite to European Paediatric Neurology Society Meeting, Baden Baden, Germany, 11 September 2001. Eur J Paediatr Neurol. 2002;6:293–297. doi: 10.1053/ejpn.2002.0612. - DOI - PubMed
-
- Archer H, Evans J, Leonard H, Colvin L, Ravine D, Christodoulou J, Williamson S, Charman T, Bailey ME, Sampson J. et al.Correlation between clinical severity in patients with Rett syndrome with a p.R168X or p.T158M MECP2 mutation, and the direction and degree of skewing of X-chromosome inactivation. J Med Genet. 2007;44:148–152. - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
