

Synthesis, biological, and antitumor activity of a highly potent 6-substituted pyrrolo[2,3-d]pyrimidine thienoyl antifolate inhibitor with proton-coupled folate transporter and folate receptor selectivity over the reduced folate carrier that inhibits β-glycinamide ribonucleotide formyltransferase
- PMID: 21879757
- PMCID: PMC3209708
- DOI: 10.1021/jm200739e
Synthesis, biological, and antitumor activity of a highly potent 6-substituted pyrrolo[2,3-d]pyrimidine thienoyl antifolate inhibitor with proton-coupled folate transporter and folate receptor selectivity over the reduced folate carrier that inhibits β-glycinamide ribonucleotide formyltransferase
Abstract
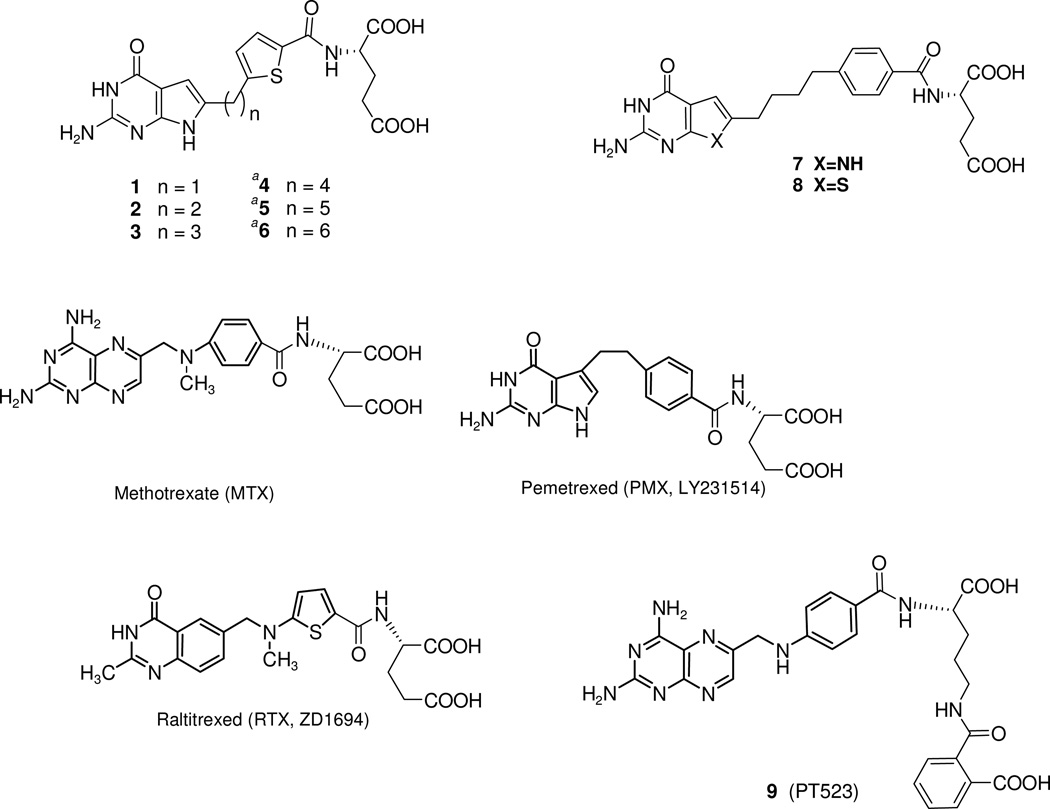
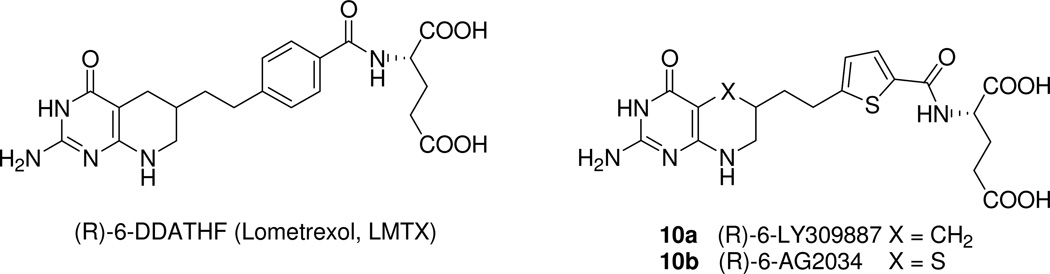
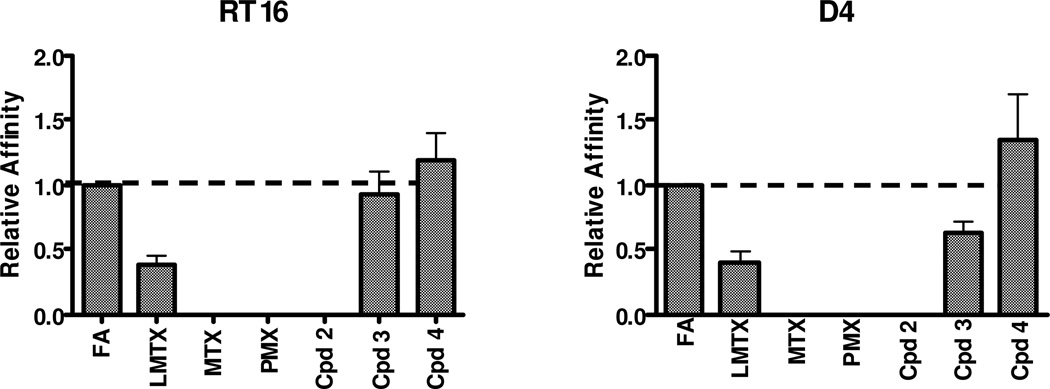
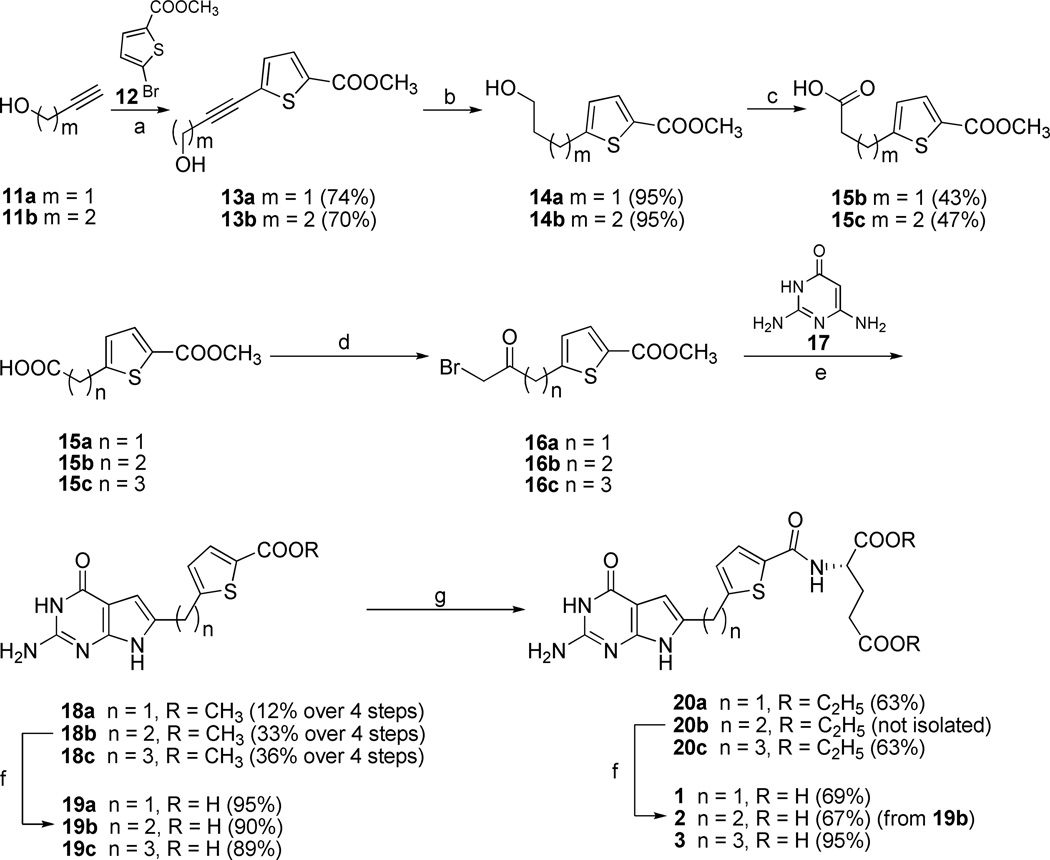
2-Amino-4-oxo-6-substituted pyrrolo[2,3-d]pyrimidine antifolates with a thienoyl side chain (compounds 1-3, respectively) were synthesized for comparison with compound 4, the previous lead compound of this series. Conversion of hydroxyl acetylen-thiophene carboxylic esters to thiophenyl-α-bromomethylketones and condensation with 2,4-diamino-6-hydroxypyrimidine afforded the 6-substituted pyrrolo[2,3-d]pyrimidine compounds of type 18 and 19. Coupling with l-glutamate diethyl ester, followed by saponification, afforded 1-3. Compound 3 selectively inhibited the proliferation of cells expressing folate receptors (FRs) α or β, or the proton-coupled folate transporter (PCFT), including KB and IGROV1 human tumor cells, much more potently than 4. Compound 3 was more inhibitory than 4 toward β-glycinamide ribonucleotide formyltransferase (GARFTase). Both 3 and 4 depleted cellular ATP pools. In SCID mice with IGROV1 tumors, 3 was more efficacious than 4. Collectively, our results show potent antitumor activity for 3 in vitro and in vivo, associated with its selective membrane transport by FRs and PCFT over RFC and inhibition of GARFTase, clearly establishing the 3-atom bridge as superior to the 1-, 2-, and 4-atom bridge lengths for the activity of this series.
Figures





Similar articles
-
6-Substituted Pyrrolo[2,3-d]pyrimidine Thienoyl Regioisomers as Targeted Antifolates for Folate Receptor α and the Proton-Coupled Folate Transporter in Human Tumors.J Med Chem. 2015 Sep 10;58(17):6938-59. doi: 10.1021/acs.jmedchem.5b00801. Epub 2015 Aug 28. J Med Chem. 2015. PMID: 26317331 Free PMC article.
-
Synthesis and antitumor activity of a novel series of 6-substituted pyrrolo[2,3-d]pyrimidine thienoyl antifolate inhibitors of purine biosynthesis with selectivity for high affinity folate receptors and the proton-coupled folate transporter over the reduced folate carrier for cellular entry.J Med Chem. 2010 Feb 11;53(3):1306-18. doi: 10.1021/jm9015729. J Med Chem. 2010. PMID: 20085328 Free PMC article.
-
Tumor Targeting with Novel 6-Substituted Pyrrolo [2,3-d] Pyrimidine Antifolates with Heteroatom Bridge Substitutions via Cellular Uptake by Folate Receptor α and the Proton-Coupled Folate Transporter and Inhibition of de Novo Purine Nucleotide Biosynthesis.J Med Chem. 2016 Sep 8;59(17):7856-76. doi: 10.1021/acs.jmedchem.6b00594. Epub 2016 Aug 26. J Med Chem. 2016. PMID: 27458733 Free PMC article.
-
The promise and challenges of exploiting the proton-coupled folate transporter for selective therapeutic targeting of cancer.Cancer Chemother Pharmacol. 2018 Jan;81(1):1-15. doi: 10.1007/s00280-017-3473-8. Epub 2017 Nov 10. Cancer Chemother Pharmacol. 2018. PMID: 29127457 Free PMC article. Review.
-
Biology of the major facilitative folate transporters SLC19A1 and SLC46A1.Curr Top Membr. 2014;73:175-204. doi: 10.1016/B978-0-12-800223-0.00004-9. Curr Top Membr. 2014. PMID: 24745983 Free PMC article. Review.
Cited by
-
The human proton-coupled folate transporter: Biology and therapeutic applications to cancer.Cancer Biol Ther. 2012 Dec;13(14):1355-73. doi: 10.4161/cbt.22020. Epub 2012 Sep 6. Cancer Biol Ther. 2012. PMID: 22954694 Free PMC article. Review.
-
Functional and mechanistic roles of the human proton-coupled folate transporter transmembrane domain 6-7 linker.Biochem J. 2016 Oct 15;473(20):3545-3562. doi: 10.1042/BCJ20160399. Epub 2016 Aug 11. Biochem J. 2016. PMID: 27514717 Free PMC article.
-
4-Substituted Thieno[3,2-d]pyrimidines as Dual-Stage Antiplasmodial Derivatives.Pharmaceuticals (Basel). 2022 Jul 1;15(7):820. doi: 10.3390/ph15070820. Pharmaceuticals (Basel). 2022. PMID: 35890119 Free PMC article.
-
Fluorine-Substituted Pyrrolo[2,3- d]Pyrimidine Analogues with Tumor Targeting via Cellular Uptake by Folate Receptor α and the Proton-Coupled Folate Transporter and Inhibition of de Novo Purine Nucleotide Biosynthesis.J Med Chem. 2018 May 10;61(9):4228-4248. doi: 10.1021/acs.jmedchem.8b00408. Epub 2018 Apr 27. J Med Chem. 2018. PMID: 29701475 Free PMC article.
-
Biology and therapeutic applications of the proton-coupled folate transporter.Expert Opin Drug Metab Toxicol. 2022 Oct;18(10):695-706. doi: 10.1080/17425255.2022.2136071. Epub 2022 Oct 20. Expert Opin Drug Metab Toxicol. 2022. PMID: 36239195 Free PMC article.
References
-
- Chabner BA, Allegra C. (2011) Antifolates. In: Chabner BA, Longo DL, editors. Cancer Chemotherapy and Biotherapy: Principles and Practice. Philadelphia, PA: Lippincott Williams and Wilkins; 2011. pp. 109–138.
-
- Matherly LH, Hou Z, Deng Y. Human Reduced Folate Carrier: Translation of Basic Biology to Cancer Etiology and Therapy. Cancer Metastasis Rev. 2007;26:111–128. - PubMed
-
- Zhao R, Goldman ID. Resistance to Antifolates. Oncogene. 2003;22:7431–7457. - PubMed
-
- Elnakat H, Ratnam M. Distribution, Functionality and Gene Regulation of Folate Receptor Isoforms: Implications in Targeted Therapy. Adv. Drug Deliv. 2004;56:1067–1084. - PubMed
-
- Qiu A, Jansen M, Sakaris A, Min SH, Chattopadhyay S, Tsai E, Sandoval C, Zhao R, Akabas MH, Goldman ID. Identification of an Intestinal Folate Transporter and the Molecular Basis for Hereditary Folate Malabsorption. Cell. 2006;127:917–928. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Chemical Information
