

Genome-wide association analysis of clinical vs. nonclinical origin provides insights into Saccharomyces cerevisiae pathogenesis
- PMID: 21880084
- PMCID: PMC3183415
- DOI: 10.1111/j.1365-294X.2011.05225.x
Genome-wide association analysis of clinical vs. nonclinical origin provides insights into Saccharomyces cerevisiae pathogenesis
Abstract
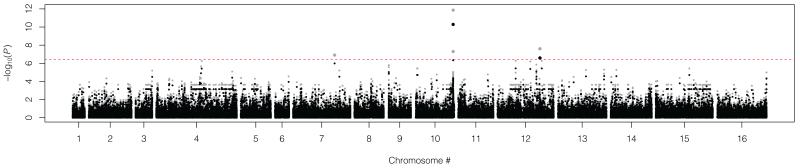
Because domesticated Saccharomyces cerevisiae strains have been used to produce fermented food and beverages for centuries without apparent health implications, S. cerevisiae has always been considered a Generally Recognized As Safe (GRAS) microorganism. However, the number of reported mucosal and systemic S. cerevisiae infections in the human population has increased and fatal infections have occurred even in relatively healthy individuals. In order to gain insight into the pathogenesis of S. cerevisiae and improve our understanding of the emergence of fungal pathogens, we performed a population-based genome-wide environmental association analysis of clinical vs. nonclinical origin in S. cerevisiae. Using tiling array-based, high-density genotypes of 44 clinical and 44 nonclinical S. cerevisiae strains from diverse geographical origins and source substrates, we identified several genetic loci associated with clinical background in S. cerevisiae. Associated polymorphisms within the coding sequences of VRP1, KIC1, SBE22 and PDR5, and the 5' upstream region of YGR146C indicate the importance of pseudohyphal formation, robust cell wall maintenance and cellular detoxification for S. cerevisiae pathogenesis, and constitute good candidates for follow-up verification of virulence and virulence-related factors underlying the pathogenicity of S. cerevisiae.
© 2011 Blackwell Publishing Ltd.
Figures










Similar articles
-
The 100-genomes strains, an S. cerevisiae resource that illuminates its natural phenotypic and genotypic variation and emergence as an opportunistic pathogen.Genome Res. 2015 May;25(5):762-74. doi: 10.1101/gr.185538.114. Epub 2015 Apr 3. Genome Res. 2015. PMID: 25840857 Free PMC article.
-
Microsatellite analysis of genetic diversity among clinical and nonclinical Saccharomyces cerevisiae isolates suggests heterozygote advantage in clinical environments.Mol Ecol. 2009 Jul;18(13):2779-86. doi: 10.1111/j.1365-294X.2009.04234.x. Epub 2009 May 20. Mol Ecol. 2009. PMID: 19457175 Free PMC article.
-
Comparison of Saccharomyces cerevisiae strains of clinical and nonclinical origin by molecular typing and determination of putative virulence traits.FEMS Yeast Res. 2008 Jun;8(4):631-40. doi: 10.1111/j.1567-1364.2008.00365.x. Epub 2008 Mar 18. FEMS Yeast Res. 2008. PMID: 18355272 Free PMC article.
-
Review of current methods for characterizing virulence and pathogenicity potential of industrial Saccharomyces cerevisiae strains towards humans.FEMS Yeast Res. 2015 Sep;15(6):fov057. doi: 10.1093/femsyr/fov057. Epub 2015 Jul 19. FEMS Yeast Res. 2015. PMID: 26195617 Review.
-
Genetic mapping of quantitative phenotypic traits in Saccharomyces cerevisiae.FEMS Yeast Res. 2012 Mar;12(2):215-27. doi: 10.1111/j.1567-1364.2011.00777.x. Epub 2012 Jan 24. FEMS Yeast Res. 2012. PMID: 22150948 Review.
Cited by
-
Genome-Wide Association for Itraconazole Sensitivity in Non-resistant Clinical Isolates of Aspergillus fumigatus.Front Fungal Biol. 2021 Jan 14;1:617338. doi: 10.3389/ffunb.2020.617338. eCollection 2020. Front Fungal Biol. 2021. PMID: 37743877 Free PMC article.
-
The 100-genomes strains, an S. cerevisiae resource that illuminates its natural phenotypic and genotypic variation and emergence as an opportunistic pathogen.Genome Res. 2015 May;25(5):762-74. doi: 10.1101/gr.185538.114. Epub 2015 Apr 3. Genome Res. 2015. PMID: 25840857 Free PMC article.
-
Whole Genome Analysis of 132 Clinical Saccharomyces cerevisiae Strains Reveals Extensive Ploidy Variation.G3 (Bethesda). 2016 Aug 9;6(8):2421-34. doi: 10.1534/g3.116.029397. G3 (Bethesda). 2016. PMID: 27317778 Free PMC article.
-
Genome-Wide Screen for Saccharomyces cerevisiae Genes Contributing to Opportunistic Pathogenicity in an Invertebrate Model Host.G3 (Bethesda). 2018 Jan 4;8(1):63-78. doi: 10.1534/g3.117.300245. G3 (Bethesda). 2018. PMID: 29122853 Free PMC article.
-
High-resolution mapping of cis-regulatory variation in budding yeast.Proc Natl Acad Sci U S A. 2017 Dec 12;114(50):E10736-E10744. doi: 10.1073/pnas.1717421114. Epub 2017 Nov 28. Proc Natl Acad Sci U S A. 2017. PMID: 29183975 Free PMC article.
References
-
- Bassetti S, Frei R, Zimmerli W. Fungemia with Saccharomyces cerevisiae after treatment with Saccharomyces boulardii. American Journal of Medicine. 1998;105:71–72. - PubMed
-
- Bengtsson H, Simpson K, Bullard J, Hansen K. Technical Report #745. Department of Statistics, University of California; Berkeley, USA: 2008. Aroma.affymetrix: a generic framework in R for analyzing small to very large Affymetrix data sets in bounded memory.
-
- Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on bias and variance. Bioinformatics. 2003;19:185–193. - PubMed
-
- Borth N, Walther A, Reijnst P, Jorde S, Schaub Y, Wendland J. Candida albicans Vrp1 is required for polarized morphogenesis and interacts with Wal1 and Myo5. Microbiology. 2010;156:2962–2969. - PubMed
-
- Clemons KV, McCusker JH, Davis RW, Stevens DA. Comparative pathogenesis of clinical and nonclinical isolates of Saccharomyces cerevisiae. Journal of Infectious Diseases. 1994;169:859–67. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
