

The human gut virome: inter-individual variation and dynamic response to diet
- PMID: 21880779
- PMCID: PMC3202279
- DOI: 10.1101/gr.122705.111
The human gut virome: inter-individual variation and dynamic response to diet
Abstract
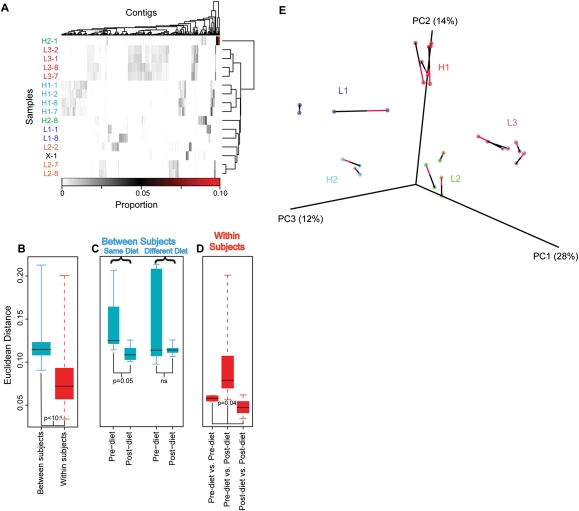
Immense populations of viruses are present in the human gut and other body sites. Understanding the role of these populations (the human "virome") in health and disease requires a much deeper understanding of their composition and dynamics in the face of environmental perturbation. Here, we investigate viromes from human subjects on a controlled feeding regimen. Longitudinal fecal samples were analyzed by metagenomic sequencing of DNA from virus-like particles (VLP) and total microbial communities. Assembly of 336 Mb of VLP sequence yielded 7175 contigs, many identifiable as complete or partial bacteriophage genomes. Contigs were rich in viral functions required in lytic and lysogenic growth, as well as unexpected functions such as viral CRISPR arrays and genes for antibiotic resistance. The largest source of variance among virome samples was interpersonal variation. Parallel deep-sequencing analysis of bacterial populations showed covaration of the virome with the larger microbiome. The dietary intervention was associated with a change in the virome community to a new state, in which individuals on the same diet converged. Thus these data provide an overview of the composition of the human gut virome and associate virome structure with diet.
Figures
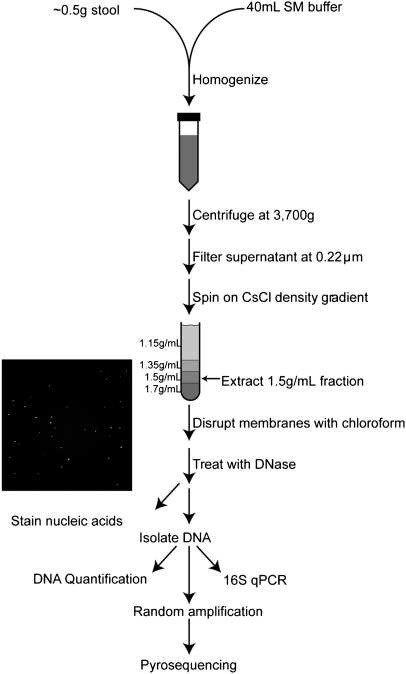
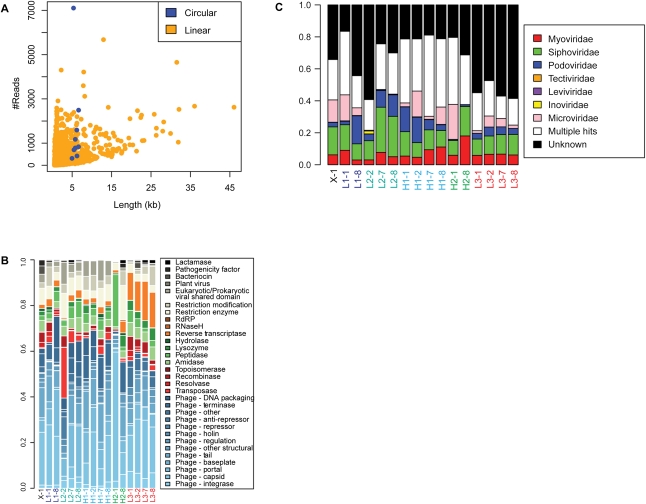
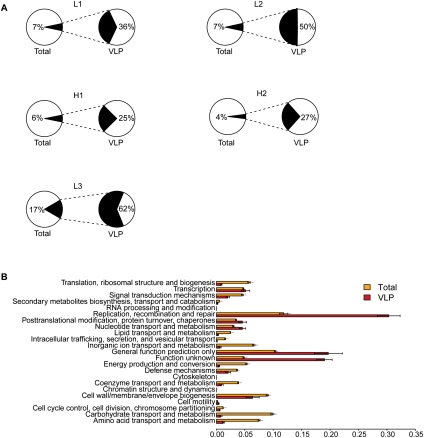
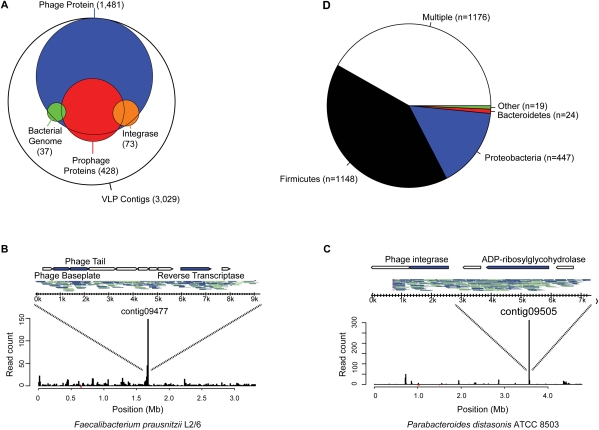
References
-
- Angly F, Rodriguez-Brito B, Bangor D, McNairnie P, Breitbart M, Salamon P, Felts B, Nulton J, Mahaffy J, Rohwer F 2005. PHACCS, an online tool for estimating the structure and diversity of uncultured viral communities using metagenomic information. BMC Bioinformatics 6: 41 doi: 10.1186/1471-2105-6-41 - PMC - PubMed
-
- Barlow M 2009. What antimicrobial resistance has taught us about horizontal gene transfer. Methods Mol Biol 532: 397–411 - PubMed
-
- Bohannan BJM, Lenski RE 1997. Effect of resource enrichment on a chemostat community of bacteria and bacteriophage. Ecology 78: 2303–2315
-
- Breitbart M, Haynes M, Kelley S, Angly F, Edwards RA, Felts B, Mahaffy JM, Mueller J, Nulton J, Rayhawk S, et al. 2008. Viral diversity and dynamics in an infant gut. Res Microbiol 159: 367–373 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous