

GnRH-deficient phenotypes in humans and mice with heterozygous variants in KISS1/Kiss1
- PMID: 21880801
- PMCID: PMC3205899
- DOI: 10.1210/jc.2011-0518
GnRH-deficient phenotypes in humans and mice with heterozygous variants in KISS1/Kiss1
Abstract
Context: KISS1 is a candidate gene for GnRH deficiency.
Objective: Our objective was to identify deleterious mutations in KISS1.
Patients and methods: DNA sequencing and assessment of the effects of rare sequence variants (RSV) were conducted in 1025 probands with GnRH-deficient conditions.
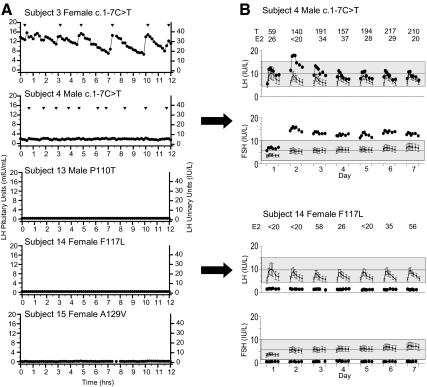
Results: Fifteen probands harbored 10 heterozygous RSV in KISS1 seen in less than 1% of control subjects. Of the variants that reside within the mature kisspeptin peptide, p.F117L (but not p.S77I, p.Q82K, p.H90D, or p.P110T) reduces inositol phosphate generation. Of the variants that lie within the coding region but outside the mature peptide, p.G35S and p.C53R (but not p.A129V) are predicted in silico to be deleterious. Of the variants that lie outside the coding region, one (g.1-3659C→T) impairs transcription in vitro, and another (c.1-7C→T) lies within the consensus Kozak sequence. Of five probands tested, four had abnormal baseline LH pulse patterns. In mice, testosterone decreases with heterozygous loss of Kiss1 and Kiss1r alleles (wild-type, 274 ± 99, to double heterozygotes, 69 ± 16 ng/dl; r(2) = 0.13; P = 0.03). Kiss1/Kiss1r double-heterozygote males have shorter anogenital distances (13.0 ± 0.2 vs. 15.6 ± 0.2 mm at P34, P < 0.001), females have longer estrous cycles (7.4 ± 0.2 vs. 5.6 ± 0.2 d, P < 0.01), and mating pairs have decreased litter frequency (0.59 ± 0.09 vs. 0.71 ± 0.06 litters/month, P < 0.04) and size (3.5 ± 0.2 vs. 5.4 ± 0.3 pups/litter, P < 0.001) compared with wild-type mice.
Conclusions: Deleterious, heterozygous RSV in KISS1 exist at a low frequency in GnRH-deficient patients as well as in the general population in presumably normal individuals. As in Kiss1(+/-)/Kiss1r(+/-) mice, heterozygous KISS1 variants in humans may work with other genetic and/or environmental factors to cause abnormal reproductive function.
Figures





References
-
- Dodé C, Levilliers J, Dupont JM, De Paepe A, Le Dû N, Soussi-Yanicostas N, Coimbra RS, Delmaghani S, Compain-Nouaille S, Baverel F, Pêcheux C, Le Tessier D, Cruaud C, Delpech M, Speleman F, Vermeulen S, Amalfitano A, Bachelot Y, Bouchard P, Cabrol S, Carel JC, Delemarre-van de Waal H, Goulet-Salmon B, Kottler ML, Richard O, Sanchez-Franco F, Saura R, Young J, Petit C, Hardelin JP. 2003. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat Genet 33:463–465 - PubMed
-
- Falardeau J, Chung WC, Beenken A, Raivio T, Plummer L, Sidis Y, Jacobson-Dickman EE, Eliseenkova AV, Ma J, Dwyer A, Quinton R, Na S, Hall JE, Huot C, Alois N, Pearce SH, Cole LW, Hughes V, Mohammadi M, Tsai P, Pitteloud N. 2008. Decreased FGF8 signaling causes deficiency of gonadotropin-releasing hormone in humans and mice. J Clin Invest 118:2822–2831 - PMC - PubMed
-
- Dodé C, Teixeira L, Levilliers J, Fouveaut C, Bouchard P, Kottler ML, Lespinasse J, Lienhardt-Roussie A, Mathieu M, Moerman A, Morgan G, Murat A, Toublanc JE, Wolczynski S, Delpech M, Petit C, Young J, Hardelin JP. 2006. Kallmann syndrome: mutations in the genes encoding prokineticin-2 and prokineticin receptor-2. PLoS Genet 2:e175. - PMC - PubMed
-
- Topaloglu AK, Reimann F, Guclu M, Yalin AS, Kotan LD, Porter KM, Serin A, Mungan NO, Cook JR, Ozbek MN, Imamoglu S, Akalin NS, Yuksel B, O'Rahilly S, Semple RK. 2009. TAC3 and TACR3 mutations in familial hypogonadotropic hypogonadism reveal a key role for Neurokinin B in the central control of reproduction. Nat Genet 41:354–358 - PMC - PubMed
-
- de Roux N, Young J, Misrahi M, Genet R, Chanson P, Schaison G, Milgrom E. 1997. A family with hypogonadotropic hypogonadism and mutations in the gonadotropin-releasing hormone receptor. N Engl J Med 337:1597–1602 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
