

Genetics of kidney failure and the evolving story of APOL1
- PMID: 21881214
- PMCID: PMC3163957
- DOI: 10.1172/JCI46263
Genetics of kidney failure and the evolving story of APOL1
Abstract
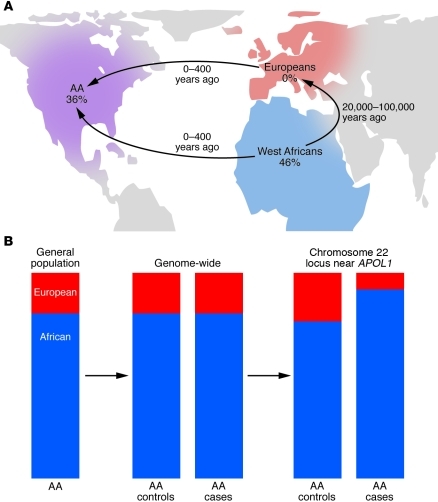
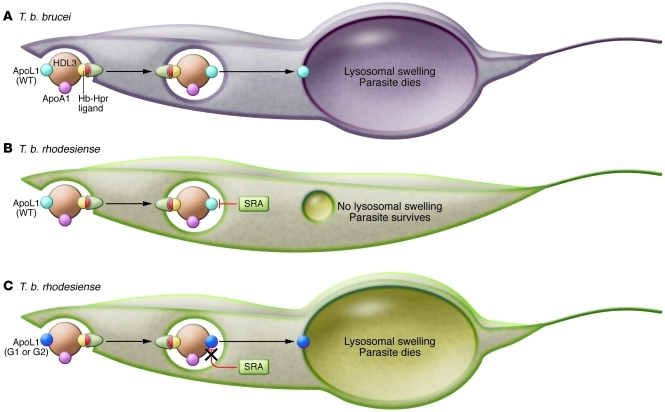
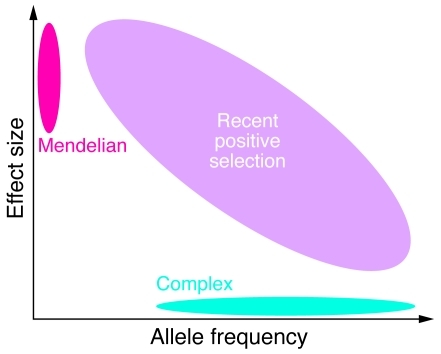
Chronic kidney disease (CKD) results from a wide array of processes that impair the kidney's ability to perform its major functions. As many as 20 million Americans suffer from CKD and nearly a half million from end-stage renal disease, but there are also examples of centenarians with adequate renal function. Family-based and genome-wide studies suggest that genetic differences substantially influence an individual's lifetime risk for kidney disease. One emerging theme is that evolution of genes related to host defense against pathogens may limit kidney longevity. The identification of these genetic factors will be critical for expanding our understanding of renal development and function as well as for the design of novel therapeutics for kidney disease.
Figures
References
-
- US Renal Data System.USRDS 2009 annual data report: atlas of chronic kidney disease and end-stage renal disease in the United States . Bethesda, Maryland, USA: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases; 2009.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
