

Innate and adaptive immune responses to cell death
- PMID: 21884177
- PMCID: PMC3170128
- DOI: 10.1111/j.1600-065X.2011.01040.x
Innate and adaptive immune responses to cell death
Abstract
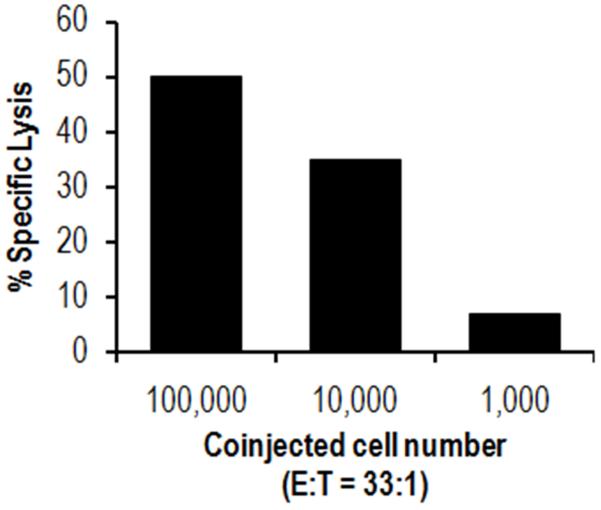
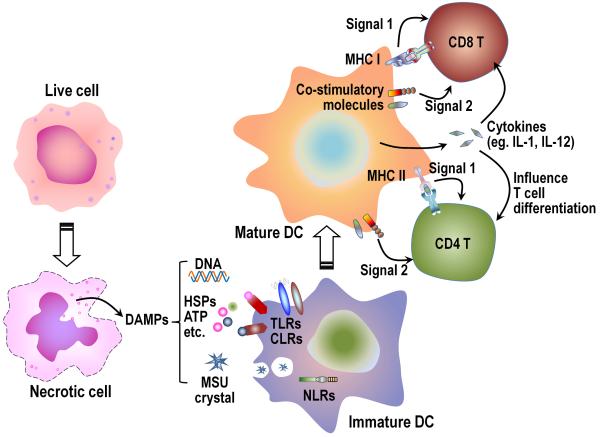
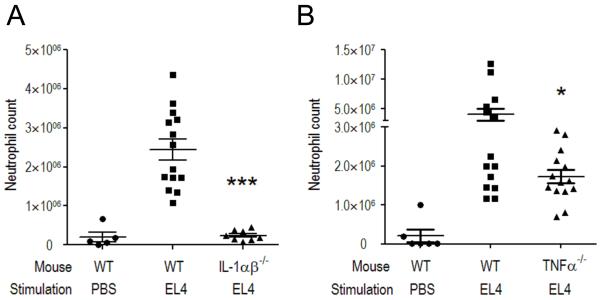
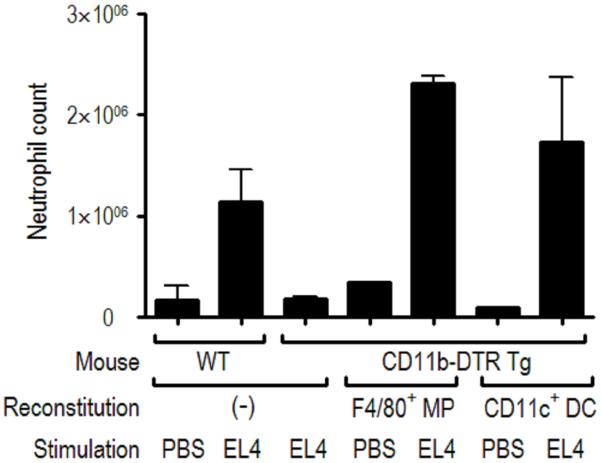
The immune system plays an essential role in protecting the host against infections and to accomplish this task has evolved mechanisms to recognize microbes and destroy them. In addition, it monitors the health of cells and responds to ones that have been injured and killed, even if this occurs under sterile conditions. This process is initiated when dying cells expose intracellular molecules that can be recognized by cells of the innate immune system. As a consequence of this recognition, dendritic cells are activated in ways that help to promote T-cell responses to antigens associated with the dying cells. In addition, macrophages are stimulated to produce the cytokine interleukin-1 that then acts on radioresistant parenchymal cells in the host in ways that drive a robust inflammatory response. In addition to dead cells, a number of other sterile particles and altered physiological states can similarly stimulate an inflammatory response and do so through common pathways involving the inflammasome and interleukin-1. These pathways underlie the pathogenesis of a number of diseases.
© 2011 John Wiley & Sons A/S.
Figures

References
-
- Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045. - PubMed
-
- Matzinger P. The danger model: a renewed sense of self. Science. 2002;296:301–305. - PubMed
-
- Majno G, Joris I. Cells, tissues, and disease: principles of general pathology. 2nd ed. Oxford University Press; New York; Oxford: 2004.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
