

Genome-Wide Gene Expression Profiling of Nucleus Accumbens Neurons Projecting to Ventral Pallidum Using both Microarray and Transcriptome Sequencing
- PMID: 21886604
- PMCID: PMC3155868
- DOI: 10.3389/fnins.2011.00098
Genome-Wide Gene Expression Profiling of Nucleus Accumbens Neurons Projecting to Ventral Pallidum Using both Microarray and Transcriptome Sequencing
Abstract
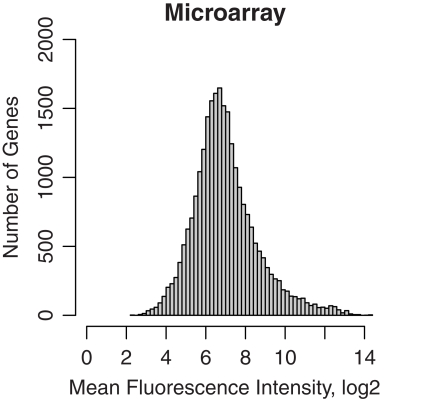
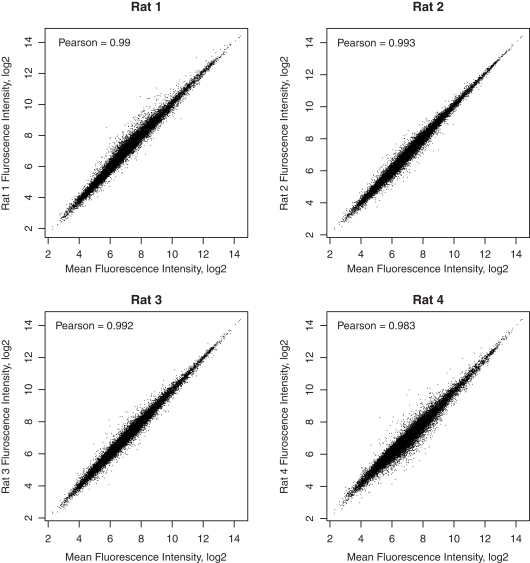
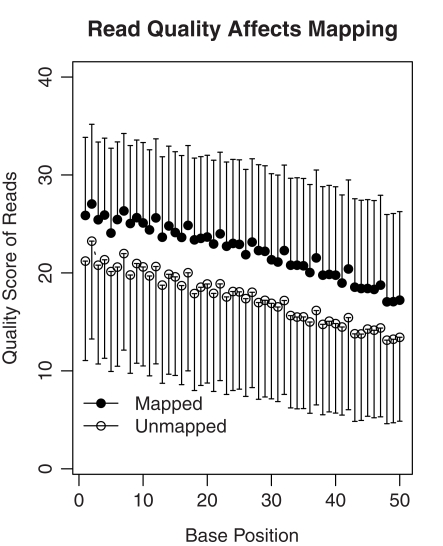
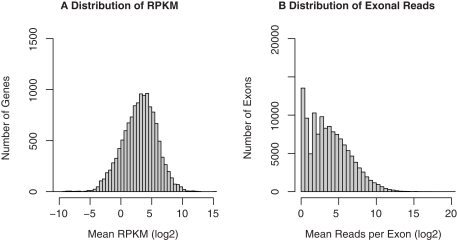
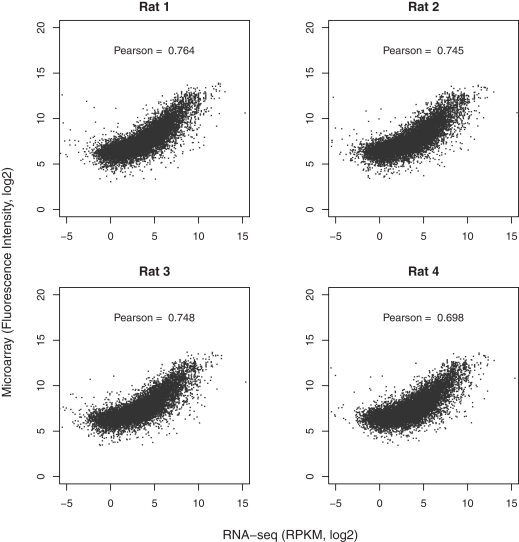
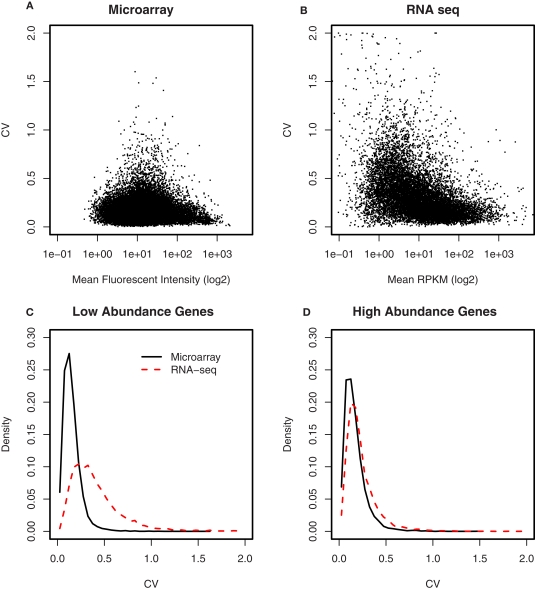
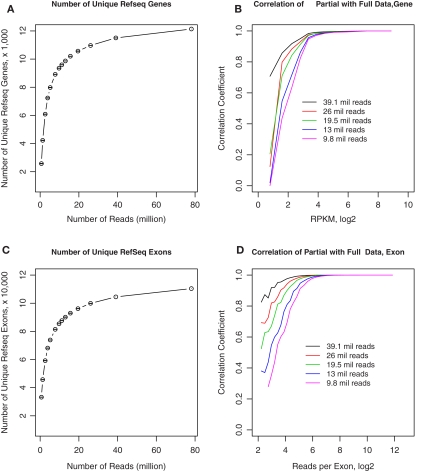
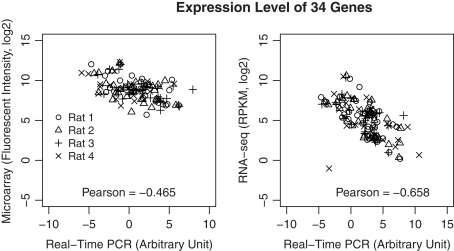
The cellular heterogeneity of brain poses a particularly thorny issue in genome-wide gene expression studies. Because laser capture microdissection (LCM) enables the precise extraction of a small area of tissue, we combined LCM with neuronal track tracing to collect nucleus accumbens shell neurons that project to ventral pallidum, which are of particular interest in the study of reward and addiction. Four independent biological samples of accumbens projection neurons were obtained. Approximately 500 pg of total RNA from each sample was then amplified linearly and subjected to Affymetrix microarray and Applied Biosystems sequencing by oligonucleotide ligation and detection (SOLiD) transcriptome sequencing (RNA-seq). A total of 375 million 50-bp reads were obtained from RNA-seq. Approximately 57% of these reads were mapped to the rat reference genome (Baylor 3.4/rn4). Approximately 11,000 unique RefSeq genes and 100,000 unique exons were identified from each sample. Of the unmapped reads, the quality scores were 4.74 ± 0.42 lower than the mapped reads. When RNA-seq and microarray data from the same samples were compared, Pearson correlations were between 0.764 and 0.798. The variances in data obtained for the four samples by microarray and RNA-seq were similar for medium to high abundance genes, but less among low abundance genes detected by microarray. Analysis of 34 genes by real-time polymerase chain reaction showed higher correlation with RNA-seq (0.66) than with microarray (0.46). Further analysis showed 20-30 million 50-bp reads are sufficient to provide estimates of gene expression levels comparable to those produced by microarray. In summary, this study showed that picogram quantities of total RNA obtained by LCM of ∼700 individual neurons is sufficient to take advantage of the benefits provided by the transcriptome sequencing technology, such as low background noise, high dynamic range, and high precision.
Keywords: GABA; GABAergic; RNA sequencing; laser capture microdissection; microarray; nucleus accumbens; transcriptome sequencing.
Figures

References
-
- Gentleman R. C., Carey V. J., Bates D. M., Bolstad B., Dettling M., Dudoit S., Ellis B., Gautier L., Ge Y., Gentry J., Hornik K., Hothorn T., Huber W., Iacus S., Irizarry R., Leisch F., Li C., Maechler M., Rossini A. J., Sawitzki G., Smith C., Smyth G., Tierney L., Yang J. Y., Zhang J. (2004). Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5, R80. 10.1186/gb-2004-5-10-r80 - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources