

Fukutin-related protein resides in the Golgi cisternae of skeletal muscle fibres and forms disulfide-linked homodimers via an N-terminal interaction
- PMID: 21886772
- PMCID: PMC3160285
- DOI: 10.1371/journal.pone.0022968
Fukutin-related protein resides in the Golgi cisternae of skeletal muscle fibres and forms disulfide-linked homodimers via an N-terminal interaction
Abstract
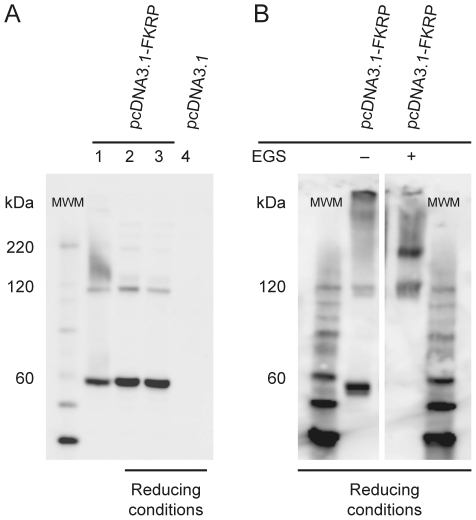
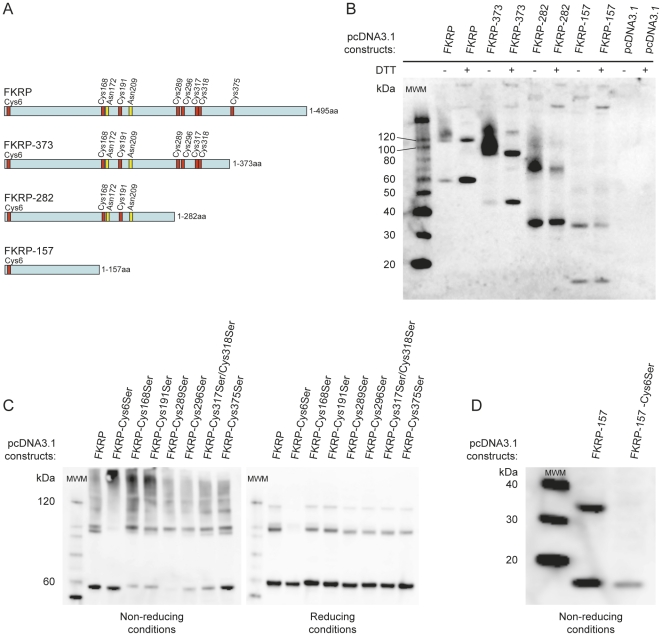
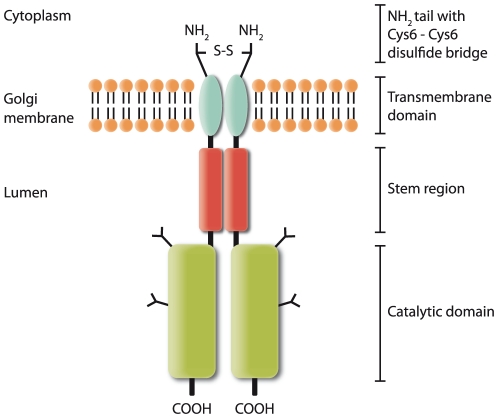
Limb-Girdle Muscular Dystrophy type 2I (LGMD2I) is an inheritable autosomal, recessive disorder caused by mutations in the FuKutin-Related Protein (FKRP) gene (FKRP) located on chromosome 19 (19q13.3). Mutations in FKRP are also associated with Congenital Muscular Dystrophy (MDC1C), Walker-Warburg Syndrome (WWS) and Muscle Eye Brain disease (MEB). These four disorders share in common an incomplete/aberrant O-glycosylation of the membrane/extracellular matrix (ECM) protein α-dystroglycan. However, further knowledge on the FKRP structure and biological function is lacking, and its intracellular location is controversial. Based on immunogold electron microscopy of human skeletal muscle sections we demonstrate that FKRP co-localises with the middle-to-trans-Golgi marker MG160, between the myofibrils in human rectus femoris muscle fibres. Chemical cross-linking experiments followed by pairwise yeast 2-hybrid experiments, and co-immune precipitation, demonstrate that FKRP can exist as homodimers as well as in large multimeric protein complexes when expressed in cell culture. The FKRP homodimer is kept together by a disulfide bridge provided by the most N-terminal cysteine, Cys6. FKRP contains N-glycan of high mannose and/or hybrid type; however, FKRP N-glycosylation is not required for FKRP homodimer or multimer formation. We propose a model for FKRP which is consistent with that of a Golgi resident type II transmembrane protein.
Conflict of interest statement
Figures



References
-
- Kanagawa M, Toda T. The genetic and molecular basis of muscular dystrophy: roles of cell-matrix linkage in the pathogenesis. J Hum Genet. 2006;51:915–926. - PubMed
-
- Barresi R, Campbell KP. Dystroglycan: from biosynthesis to pathogenesis of human disease. J Cell Sci. 2006;119:199–207. - PubMed
-
- Henry MD, Campbell KP. Dystroglycan inside and out. Curr Opin Cell Biol. 1999;11:602–607. - PubMed
-
- Ervasti JM, Campbell KP. Membrane organization of the dystrophin- glycoprotein complex. Cell. 1991;66:1121–1131. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
