

Immunoexcitotoxicity as a central mechanism in chronic traumatic encephalopathy-A unifying hypothesis
- PMID: 21886880
- PMCID: PMC3157093
- DOI: 10.4103/2152-7806.83391
Immunoexcitotoxicity as a central mechanism in chronic traumatic encephalopathy-A unifying hypothesis
Abstract
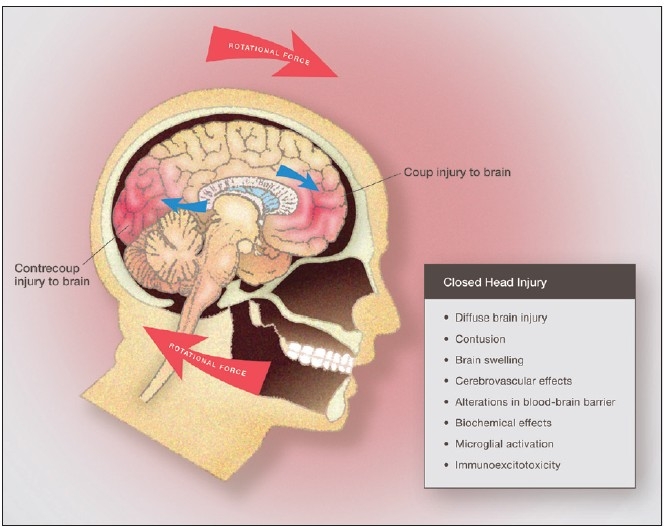
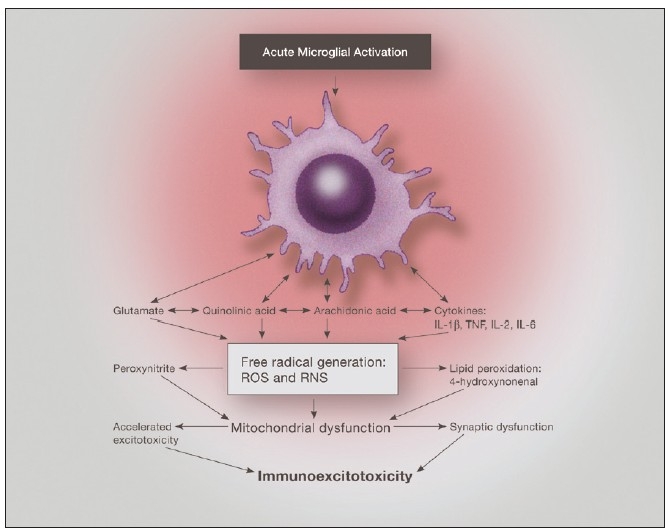
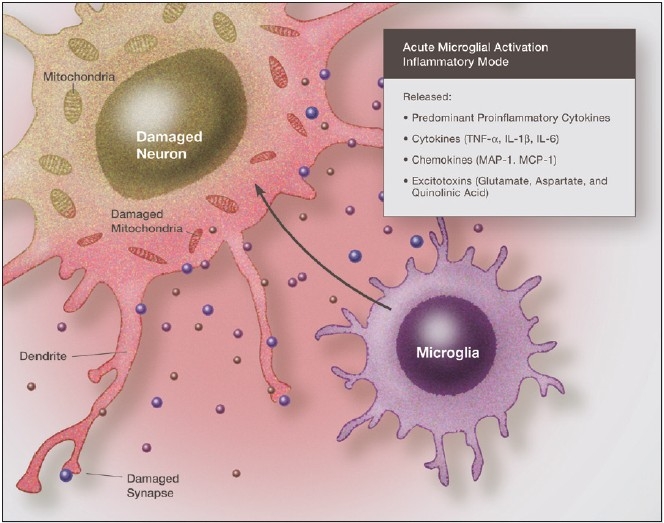
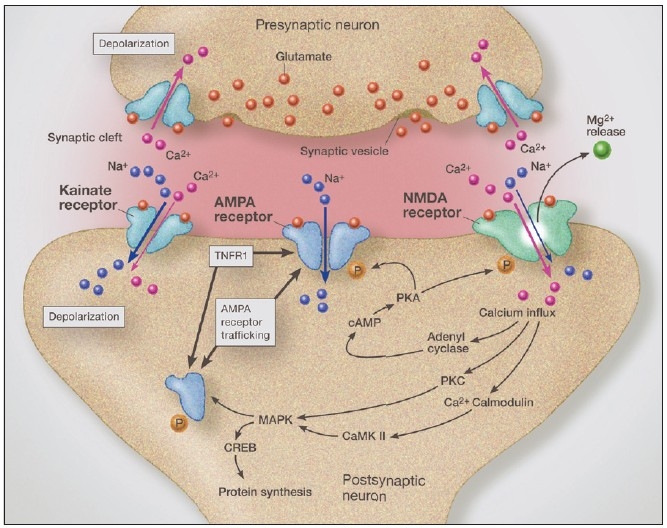
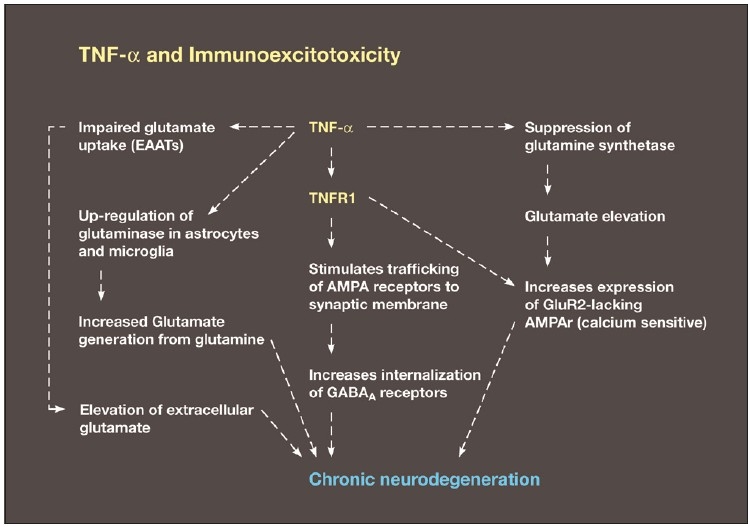
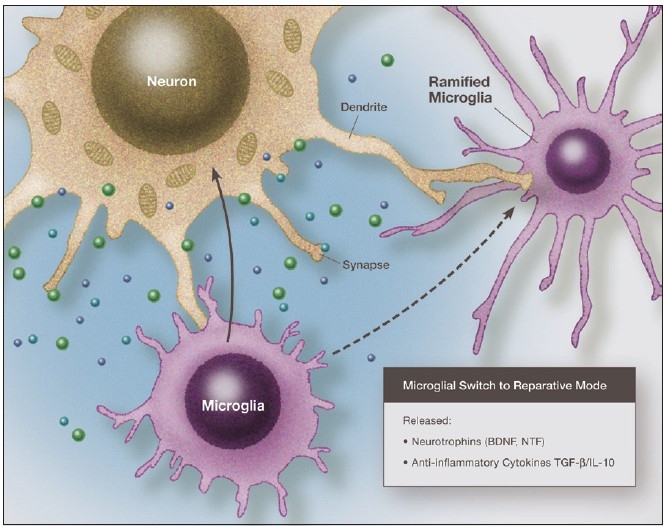
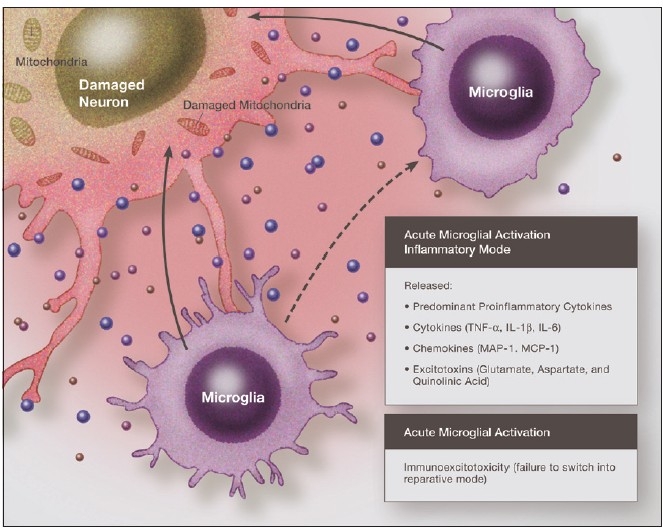
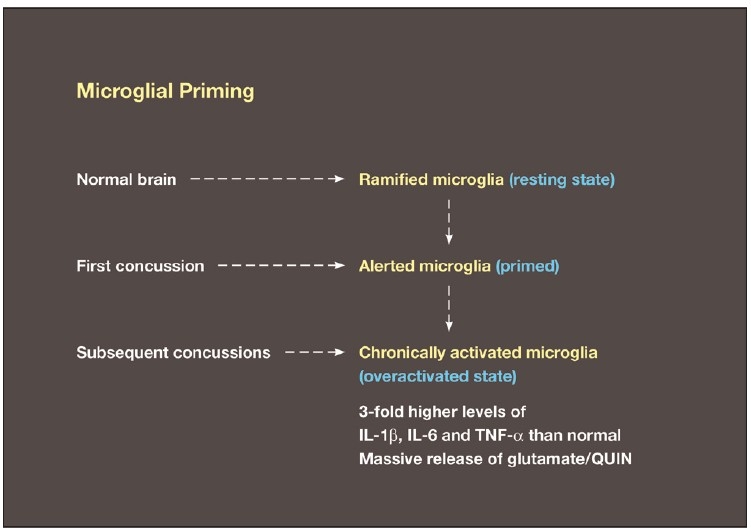
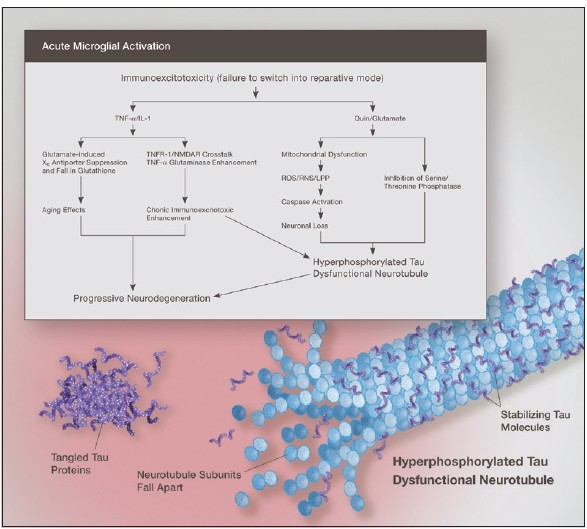
Some individuals suffering from mild traumatic brain injuries, especially repetitive mild concussions, are thought to develop a slowly progressive encephalopathy characterized by a number of the neuropathological elements shared with various neurodegenerative diseases. A central pathological mechanism explaining the development of progressive neurodegeneration in this subset of individuals has not been elucidated. Yet, a large number of studies indicate that a process called immunoexcitotoxicity may be playing a central role in many neurodegenerative diseases including chronic traumatic encephalopathy (CTE). The term immunoexcitotoxicity was first coined by the lead author to explain the evolving pathological and neurodevelopmental changes in autism and the Gulf War Syndrome, but it can be applied to a number of neurodegenerative disorders. The interaction between immune receptors within the central nervous system (CNS) and excitatory glutamate receptors trigger a series of events, such as extensive reactive oxygen species/reactive nitrogen species generation, accumulation of lipid peroxidation products, and prostaglandin activation, which then leads to dendritic retraction, synaptic injury, damage to microtubules, and mitochondrial suppression. In this paper, we discuss the mechanism of immunoexcitotoxicity and its link to each of the pathophysiological and neurochemical events previously described with CTE, with special emphasis on the observed accumulation of hyperphosphorylated tau.
Keywords: Cerebral concussion; chronic traumatic encephalopathy; cytokines; hyperphosphorylated tau; immunoexcitotoxicity; microglia; mild traumatic brain injury; quniolinic acid.
Figures

References
-
- Adams JH, Doyle D, Ford I, Gennarelli TA, Graham DI, McClellan DR. Diffuse axonal injury in head injury: definition, diagnosis and grading. Histopathology. 1989;15:49–59. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
