

Laforin, a dual specificity phosphatase involved in Lafora disease, is present mainly as monomeric form with full phosphatase activity
- PMID: 21887368
- PMCID: PMC3162602
- DOI: 10.1371/journal.pone.0024040
Laforin, a dual specificity phosphatase involved in Lafora disease, is present mainly as monomeric form with full phosphatase activity
Abstract
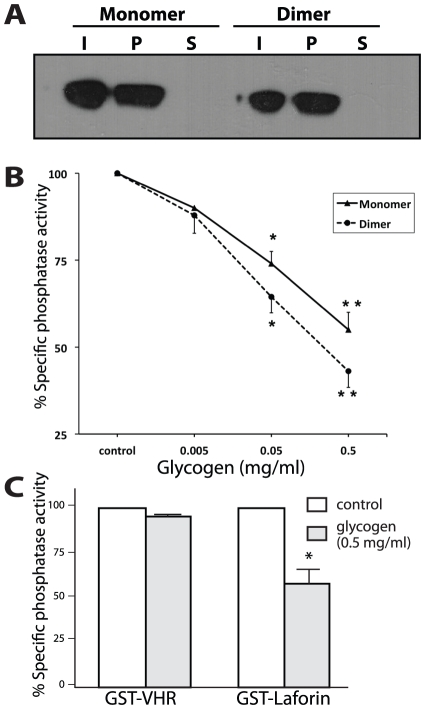
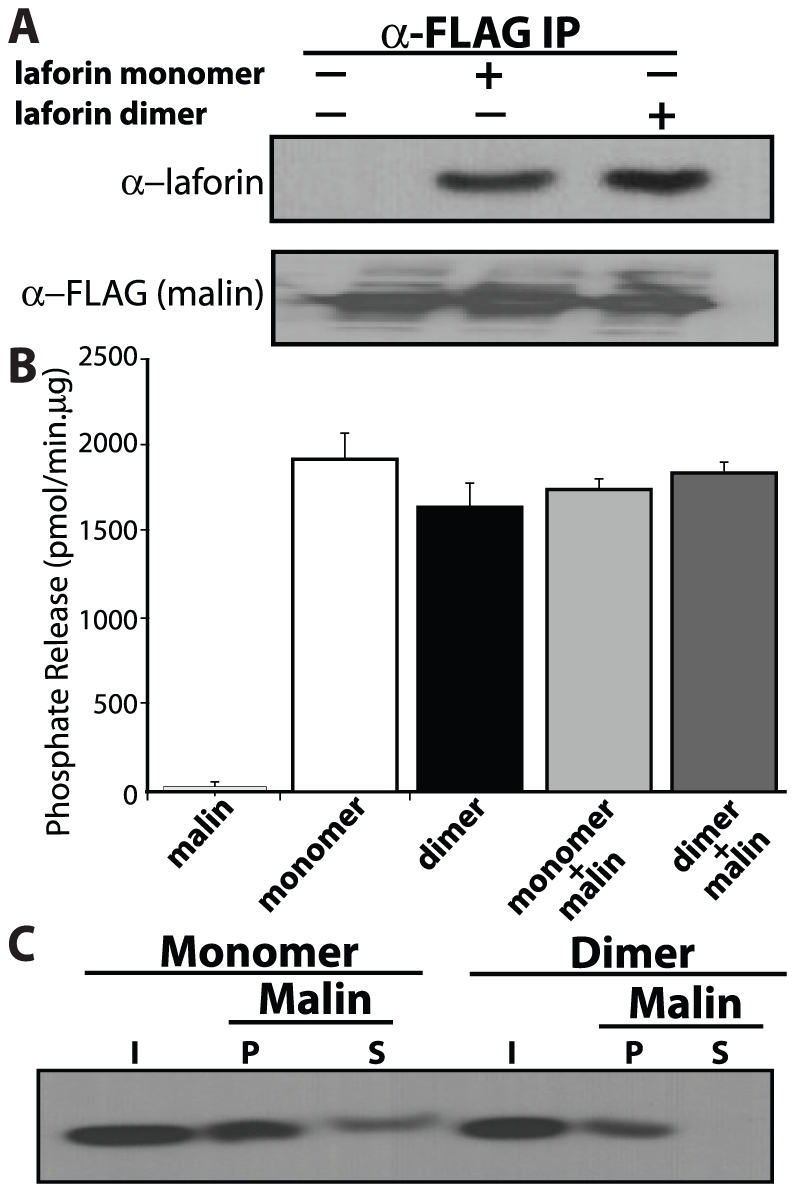
Lafora Disease (LD) is a fatal neurodegenerative epileptic disorder that presents as a neurological deterioration with the accumulation of insoluble, intracellular, hyperphosphorylated carbohydrates called Lafora bodies (LBs). LD is caused by mutations in either the gene encoding laforin or malin. Laforin contains a dual specificity phosphatase domain and a carbohydrate-binding module, and is a member of the recently described family of glucan phosphatases. In the current study, we investigated the functional and physiological relevance of laforin dimerization. We purified recombinant human laforin and subjected the monomer and dimer fractions to denaturing gel electrophoresis, mass spectrometry, phosphatase assays, protein-protein interaction assays, and glucan binding assays. Our results demonstrate that laforin prevalently exists as a monomer with a small dimer fraction both in vitro and in vivo. Of mechanistic importance, laforin monomer and dimer possess equal phosphatase activity, and they both associate with malin and bind glucans to a similar extent. However, we found differences between the two states' ability to interact simultaneously with malin and carbohydrates. Furthermore, we tested other members of the glucan phosphatase family. Cumulatively, our data suggest that laforin monomer is the dominant form of the protein and that it contains phosphatase activity.
Conflict of interest statement
Figures
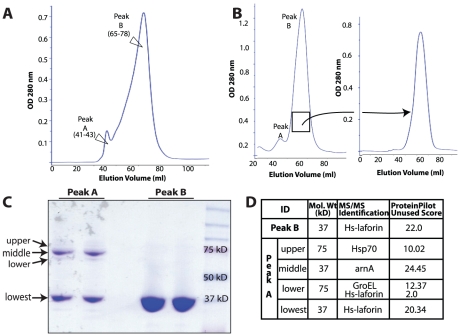
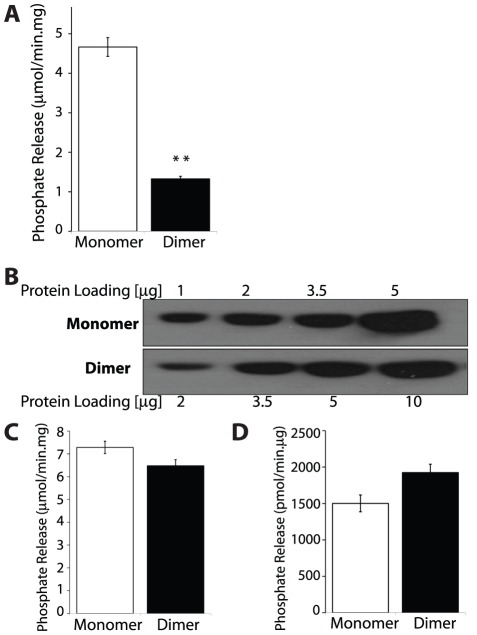
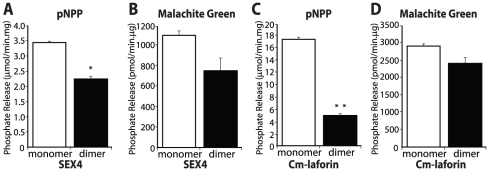
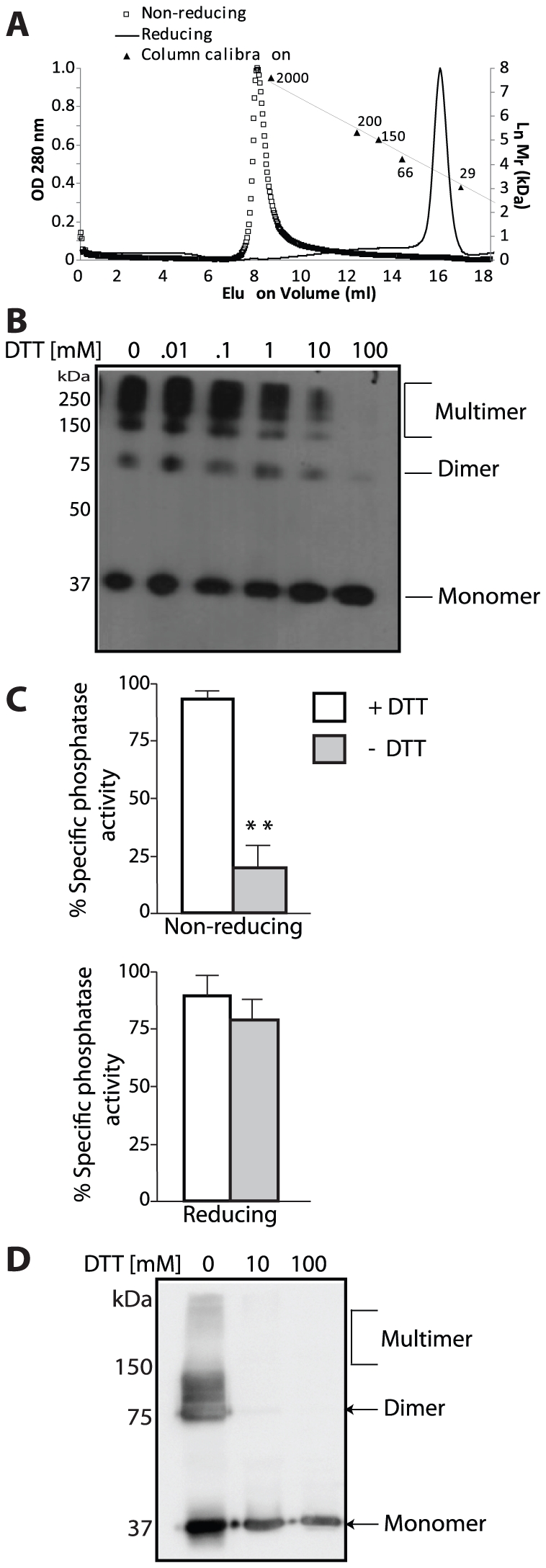
References
-
- Minassian BA, Lee JR, Herbrick JA, Huizenga J, Soder S, et al. Mutations in a gene encoding a novel protein tyrosine phosphatase cause progressive myoclonus epilepsy. Nat Genet. 1998;20:171–174. - PubMed
-
- Serratosa JM, Gomez-Garre P, Gallardo ME, Anta B, de Bernabe DB, et al. A novel protein tyrosine phosphatase gene is mutated in progressive myoclonus epilepsy of the Lafora type (EPM2). Hum Mol Genet. 1999;8:345–352. - PubMed
-
- Berkovic SF, So NK, Andermann F. Progressive myoclonus epilepsies: clinical and neurophysiological diagnosis. J Clin Neurophysiol. 1991;8:261–274. - PubMed
-
- Janeway R, Ravens JR, Pearce LA, Odor DL, Suzuki K. Progressive myoclonus epilepsy with Lafora inclusion bodies. I. Clinical, genetic, histopathologic, and biochemical aspects. Arch Neurol. 1967;16:565–582. - PubMed
-
- Van Heycop Ten Ham MW. Lafora disease, a form of progressive myoclonus epilepsy. In: Vinken PJ, Bruyn GW, editors. Handbook of Clinical neurology. Amsterdam, Holland: North Holland Publishing Company; 1975. pp. 382–422.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
