

Markers of severe vaso-occlusive painful episode frequency in children and adolescents with sickle cell anemia
- PMID: 21890147
- PMCID: PMC3258348
- DOI: 10.1016/j.jpeds.2011.07.018
Markers of severe vaso-occlusive painful episode frequency in children and adolescents with sickle cell anemia
Abstract
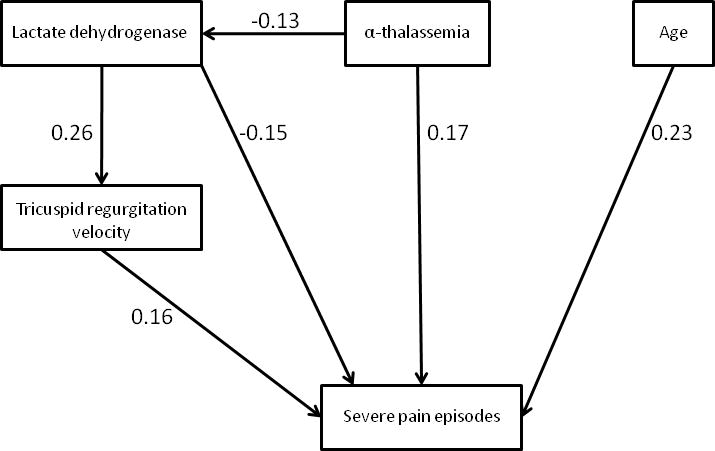
Objective: To identify factors associated with frequent severe vaso-occlusive pain crises in a contemporary pediatric cohort of patients with sickle cell anemia (SCA) enrolled in a prospective study of pulmonary hypertension and the hypoxic response in sickle cell disease.
Study design: Clinical and laboratory characteristics of children with SCA who had ≥3 severe pain crises requiring health care in the preceding year were compared with those of subjects with <3 such episodes.
Results: Seventy-five children (20%) reported ≥3 severe pain episodes in the preceding year, and 232 (61%) had none. Frequent pain episodes were associated with older age (OR, 1.2; 95% CI, 1.1-1.3; P < .0001), α-thalassemia trait (OR 3.5; 1.6-6.7; P = .002), higher median hemoglobin (OR 1.7; 95% CI: 1.2-2.4; P < .003), and lower lactate dehydrogenase concentration (OR 1.82; 95% CI: 1.07-3.11; P = .027). Children with high pain frequency also had an increased iron burden (serum ferritin, 480 vs 198 μg/L; P = .006) and higher median tricuspid regurgitation jet velocity (2.41 vs 2.31 m/s; P = .001). Neither hydroxyurea use nor fetal hemoglobin levels were significantly different according to severe pain history.
Conclusions: In our cohort of children with SCA, increasing age was associated with higher frequency of severe pain episodes as were α-thalassemia, iron overload, higher hemoglobin and lower lactate dehydrogenase concentration, and higher tricuspid regurgitation velocity.
Trial registration: ClinicalTrials.gov NCT00495638.
Copyright © 2012 Mosby, Inc. All rights reserved.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
-
- Platt OS, Thorington BD, Brambilla DJ, Milner PF, Rosse WF, Vichinsky E, et al. Pain in sickle cell disease. Rates and risk factors. N Engl J Med. 1991;325:11–6. - PubMed
-
- Smith WR, Penberthy LT, Bovbjerg VE, McClish DK, Roberts JD, Dahman B, et al. Daily assessment of pain in adults with sickle cell disease. Ann Intern Med. 2008;148:94–101. - PubMed
-
- Baum KF, Dunn DT, Maude GH, Serjeant GR. The painful crisis of homozygous sickle cell disease. A study of the risk factors. Arch Intern Med. 1987;147:1231–4. - PubMed
-
- Billett HH, Nagel RL, Fabry ME. Paradoxical increase of painful crises in sickle cell patients with alpha-thalassemia. Blood. 1995;86:4382. - PubMed
-
- Gill FM, Sleeper LA, Weiner SJ, Brown AK, Bellevue R, Grover R, et al. Clinical events in the first decade in a cohort of infants with sickle cell disease. Cooperative Study of Sickle Cell Disease. Blood. 1995;86:776–83. - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
