

A valid mouse model of AGRIN-associated congenital myasthenic syndrome
- PMID: 21890498
- PMCID: PMC3209832
- DOI: 10.1093/hmg/ddr396
A valid mouse model of AGRIN-associated congenital myasthenic syndrome
Abstract
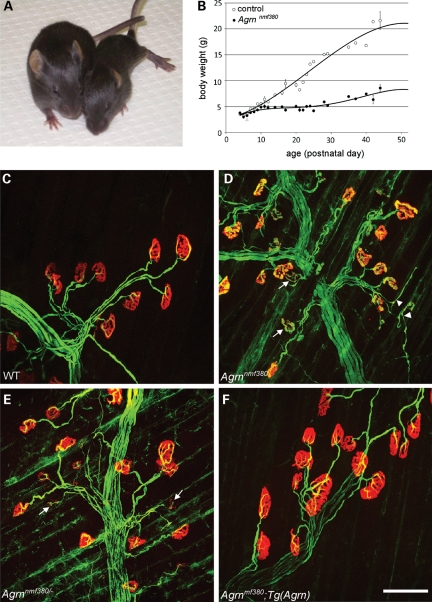
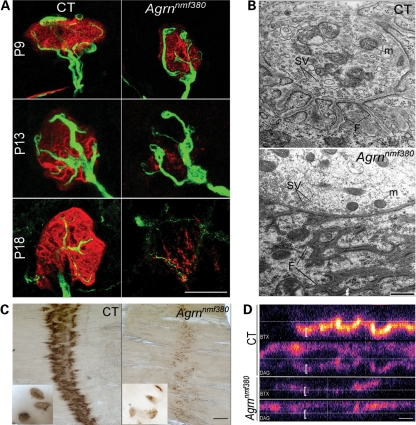
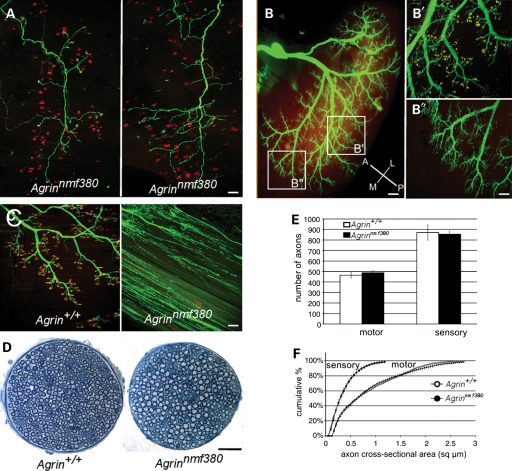
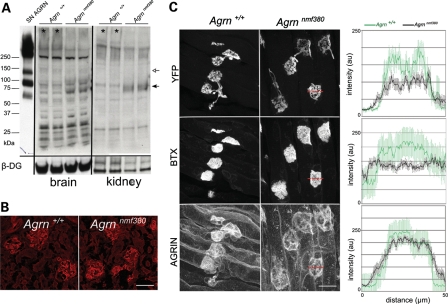
Congenital myasthenic syndromes (CMS) are inherited diseases affecting the neuromuscular junction (NMJ). Mutations in AGRIN (AGRN) and other genes in the AGRIN signaling pathway cause CMS, and gene targeting studies in mice confirm the importance of this pathway for NMJ formation. However, these mouse mutations are complete loss-of-function alleles that result in an embryonic failure of NMJ formation, and homozygous mice do not survive postpartum. Therefore, mouse models of AGRIN-related CMS that would allow preclinical testing or studies of postnatal disease progression are lacking. Using chemical mutagenesis in mice, we identified a point mutation in Agrn that results in a partial loss-of-function allele, creating a valid model of CMS. The mutation changes phenylalanine 1061 to serine in the SEA domain of AGRIN, a poorly characterized motif shared by other extracellular proteoglycans. NMJs in homozygous mice progressively degrade postnataly. Severity differs with genetic background, in different muscles, and in different regions within a muscle in a pattern matching mouse models of motor neuron disease. Mutant NMJs have decreased acetylcholine receptor density and an increased subsynaptic reticulum, evident by electron microscopy. Synapses eventually denervate and the muscles atrophy. Molecularly, several factors contribute to the partial loss of AGRIN's function. The mutant protein is found at NMJs, but is processed differently than wild-type, with decreased glycosylation, changes in sensitivity to the protease neurotrypsin and other proteolysis, and less efficient externalization and secretion. Therefore, the Agrn point mutation is a model for CMS caused by Agrn mutations and potentially other related neuromuscular diseases.
Figures




Similar articles
-
LG2 agrin mutation causing severe congenital myasthenic syndrome mimics functional characteristics of non-neural (z-) agrin.Hum Genet. 2012 Jul;131(7):1123-35. doi: 10.1007/s00439-011-1132-4. Epub 2011 Dec 29. Hum Genet. 2012. PMID: 22205389 Free PMC article.
-
Identification of an agrin mutation that causes congenital myasthenia and affects synapse function.Am J Hum Genet. 2009 Aug;85(2):155-67. doi: 10.1016/j.ajhg.2009.06.015. Epub 2009 Jul 23. Am J Hum Genet. 2009. PMID: 19631309 Free PMC article.
-
Novel SEA and LG2 Agrin mutations causing congenital Myasthenic syndrome.Orphanet J Rare Dis. 2017 Dec 19;12(1):182. doi: 10.1186/s13023-017-0732-z. Orphanet J Rare Dis. 2017. PMID: 29258548 Free PMC article.
-
Synaptic basal lamina-associated congenital myasthenic syndromes.Ann N Y Acad Sci. 2012 Dec;1275:36-48. doi: 10.1111/j.1749-6632.2012.06807.x. Ann N Y Acad Sci. 2012. PMID: 23278576 Review.
-
[Molecular mechanisms underlying the formation of neuromuscular junction].Brain Nerve. 2011 Jul;63(7):649-55. Brain Nerve. 2011. PMID: 21747134 Review. Japanese.
Cited by
-
The MuSK activator agrin has a separate role essential for postnatal maintenance of neuromuscular synapses.Proc Natl Acad Sci U S A. 2014 Nov 18;111(46):16556-61. doi: 10.1073/pnas.1408409111. Epub 2014 Nov 3. Proc Natl Acad Sci U S A. 2014. PMID: 25368159 Free PMC article.
-
Injection of a soluble fragment of neural agrin (NT-1654) considerably improves the muscle pathology caused by the disassembly of the neuromuscular junction.PLoS One. 2014 Feb 10;9(2):e88739. doi: 10.1371/journal.pone.0088739. eCollection 2014. PLoS One. 2014. PMID: 24520420 Free PMC article.
-
A Novel Egr-1-Agrin Pathway and Potential Implications for Regulation of Synaptic Physiology and Homeostasis at the Neuromuscular Junction.Front Aging Neurosci. 2017 Aug 3;9:258. doi: 10.3389/fnagi.2017.00258. eCollection 2017. Front Aging Neurosci. 2017. PMID: 28824419 Free PMC article.
-
Urinary protein biomarkers based on LC-MS/MS analysis to discriminate vascular dementia from Alzheimer's disease in Han Chinese population.Front Aging Neurosci. 2023 Jan 25;15:1070854. doi: 10.3389/fnagi.2023.1070854. eCollection 2023. Front Aging Neurosci. 2023. PMID: 36761180 Free PMC article.
-
LG2 agrin mutation causing severe congenital myasthenic syndrome mimics functional characteristics of non-neural (z-) agrin.Hum Genet. 2012 Jul;131(7):1123-35. doi: 10.1007/s00439-011-1132-4. Epub 2011 Dec 29. Hum Genet. 2012. PMID: 22205389 Free PMC article.
References
-
- Engel A.G., Ohno K., Sine S.M. Chapter 66: Congenital Myasthenic Syndrome. In: Engel A.G., Franzini-Armstrong C., editors. Myology. 3rd edn. Vol. 2. New York: McGraw-Hill; 2004. pp. 1801–1854.
-
- Sanes J.R., Lichtman J.W. Development of the vertebrate neuromuscular junction. Ann. Rev. Neurosci. 1999;22:389–442. doi:10.1146/annurev.neuro.22.1.389. - DOI - PubMed
-
- Burden S.J. Building the vertebrate neuromuscular synapse. J. Neurobiol. 2002;53:501–511. doi:10.1002/neu.10137. - DOI - PubMed
-
- Kummer T.T., Misgeld T., Sanes J.R. Assembly of the postsynaptic membrane at the neuromuscular junction: paradigm lost. Curr. Opin. Neurobiol. 2006;16:74–82. doi:10.1016/j.conb.2005.12.003. - DOI - PubMed
-
- Nitkin R.M., Smith M.A., Magill C., Fallon J.R., Yao Y.M., Wallace B.G., McMahan U.J. Identification of agrin, a synaptic organizing protein from Torpedo electric organ. J. Cell Biol. 1987;105:2471–2478. doi:10.1083/jcb.105.6.2471. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
