

Genome-wide analysis distinguishes hyperglycemia regulated epigenetic signatures of primary vascular cells
- PMID: 21890681
- PMCID: PMC3202278
- DOI: 10.1101/gr.116095.110
Genome-wide analysis distinguishes hyperglycemia regulated epigenetic signatures of primary vascular cells
Abstract
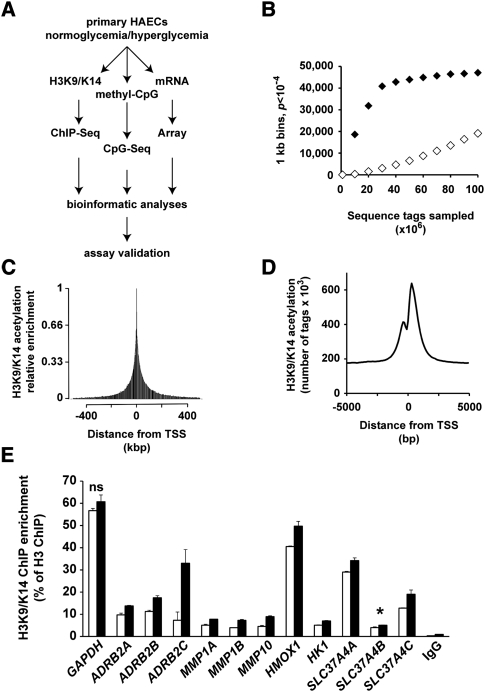
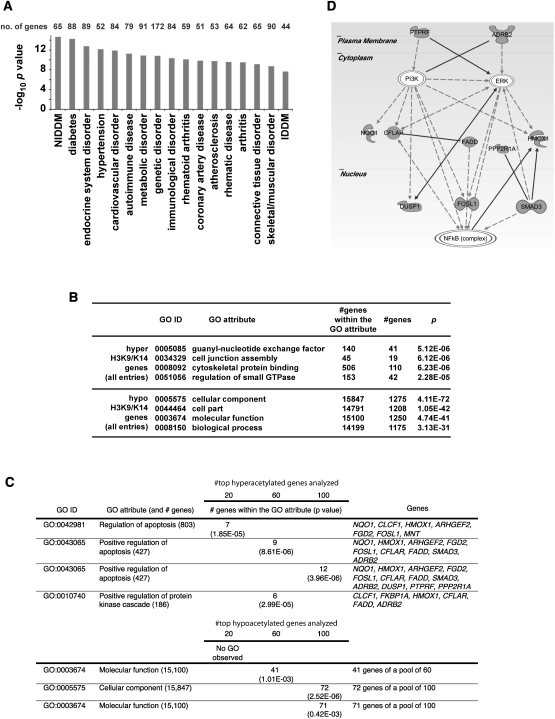
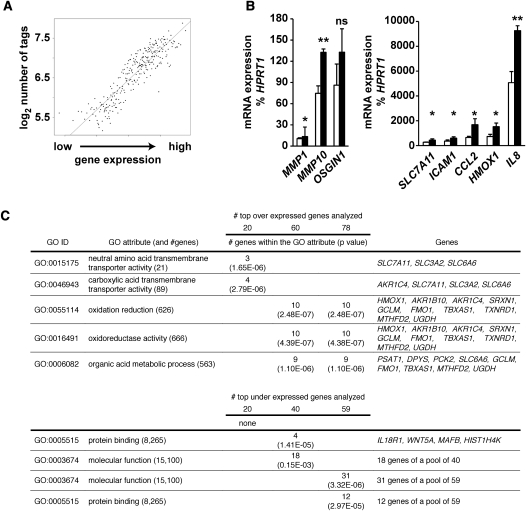
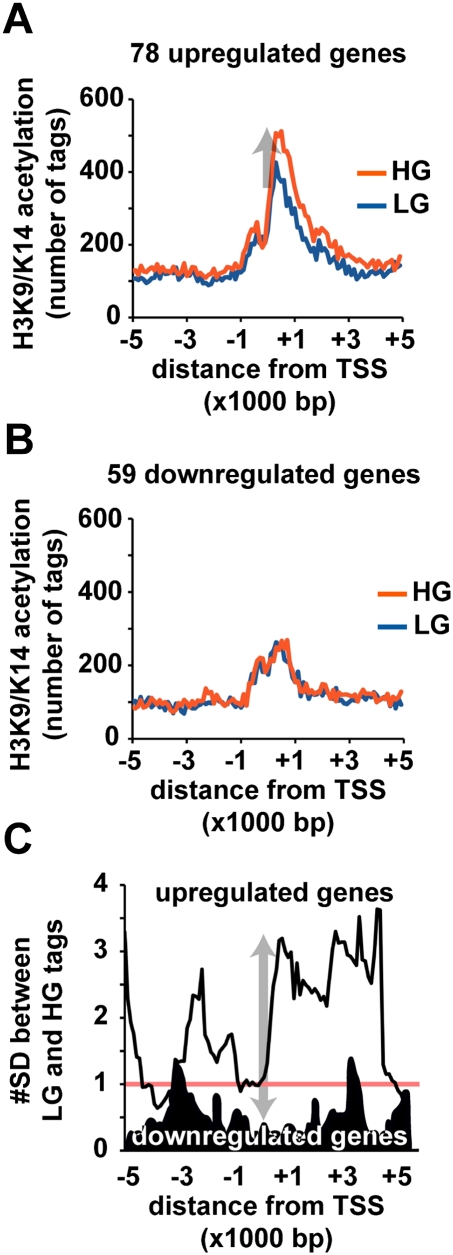
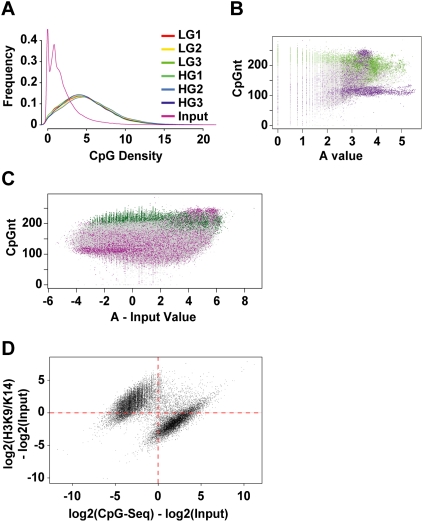
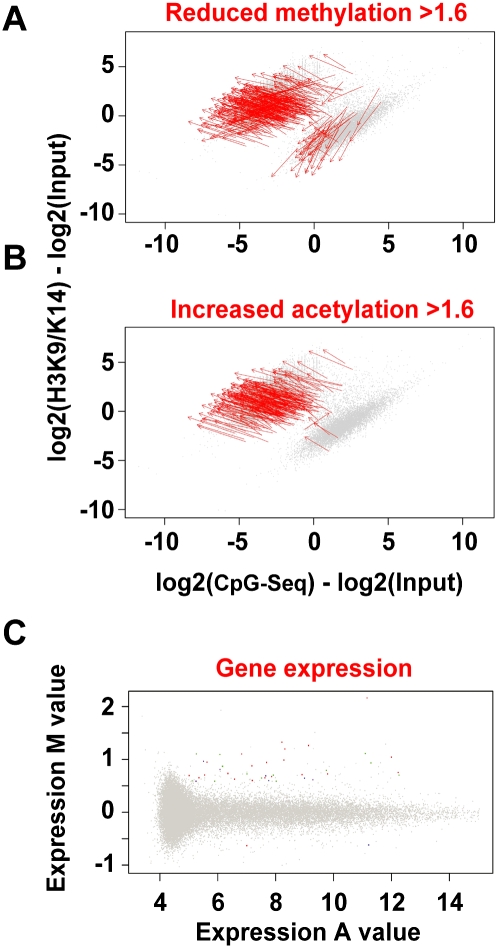
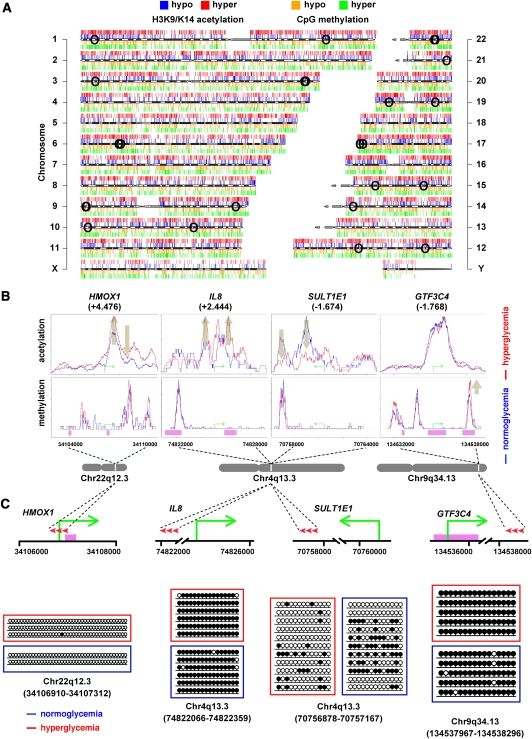
Emerging evidence suggests that poor glycemic control mediates post-translational modifications to the H3 histone tail. We are only beginning to understand the dynamic role of some of the diverse epigenetic changes mediated by hyperglycemia at single loci, yet elevated glucose levels are thought to regulate genome-wide changes, and this still remains poorly understood. In this article we describe genome-wide histone H3K9/K14 hyperacetylation and DNA methylation maps conferred by hyperglycemia in primary human vascular cells. Chromatin immunoprecipitation (ChIP) as well as CpG methylation (CpG) assays, followed by massive parallel sequencing (ChIP-seq and CpG-seq) identified unique hyperacetylation and CpG methylation signatures with proximal and distal patterns of regionalization associative with gene expression. Ingenuity knowledge-based pathway and gene ontology analyses indicate that hyperglycemia significantly affects human vascular chromatin with the transcriptional up-regulation of genes involved in metabolic and cardiovascular disease. We have generated the first installment of a reference collection of hyperglycemia-induced chromatin modifications using robust and reproducible platforms that allow parallel sequencing-by-synthesis of immunopurified content. We uncover that hyperglycemia-mediated induction of genes and pathways associated with endothelial dysfunction occur through modulation of acetylated H3K9/K14 inversely correlated with methyl-CpG content.
Figures
References
-
- Bock C, Reither S, Mikeska T, Paulsen M, Walter J, Lengauer T 2005. BiQ Analyzer: visualization and quality control for DNA methylation data from bisulfite sequencing. Bioinformatics 21: 4067–4068 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases