

Evolution of the deaminase fold and multiple origins of eukaryotic editing and mutagenic nucleic acid deaminases from bacterial toxin systems
- PMID: 21890906
- PMCID: PMC3239186
- DOI: 10.1093/nar/gkr691
Evolution of the deaminase fold and multiple origins of eukaryotic editing and mutagenic nucleic acid deaminases from bacterial toxin systems
Abstract
The deaminase-like fold includes, in addition to nucleic acid/nucleotide deaminases, several catalytic domains such as the JAB domain, and others involved in nucleotide and ADP-ribose metabolism. Using sensitive sequence and structural comparison methods, we develop a comprehensive natural classification of the deaminase-like fold and show that its ancestral version was likely to operate on nucleotides or nucleic acids. Consequently, we present evidence that a specific group of JAB domains are likely to possess a DNA repair function, distinct from the previously known deubiquitinating peptidase activity. We also identified numerous previously unknown clades of nucleic acid deaminases. Using inference based on contextual information, we suggest that most of these clades are toxin domains of two distinct classes of bacterial toxin systems, namely polymorphic toxins implicated in bacterial interstrain competition and those that target distantly related cells. Genome context information suggests that these toxins might be delivered via diverse secretory systems, such as Type V, Type VI, PVC and a novel PrsW-like intramembrane peptidase-dependent mechanism. We propose that certain deaminase toxins might be deployed by diverse extracellular and intracellular pathogens as also endosymbionts as effectors targeting nucleic acids of host cells. Our analysis suggests that these toxin deaminases have been acquired by eukaryotes on several independent occasions and recruited as organellar or nucleo-cytoplasmic RNA modifiers, operating on tRNAs, mRNAs and short non-coding RNAs, and also as mutators of hyper-variable genes, viruses and selfish elements. This scenario potentially explains the origin of mutagenic AID/APOBEC-like deaminases, including novel versions from Caenorhabditis, Nematostella and diverse algae and a large class of fast-evolving fungal deaminases. These observations greatly expand the distribution of possible unidentified mutagenic processes catalyzed by nucleic acid deaminases.
Published by Oxford University Press.
Figures

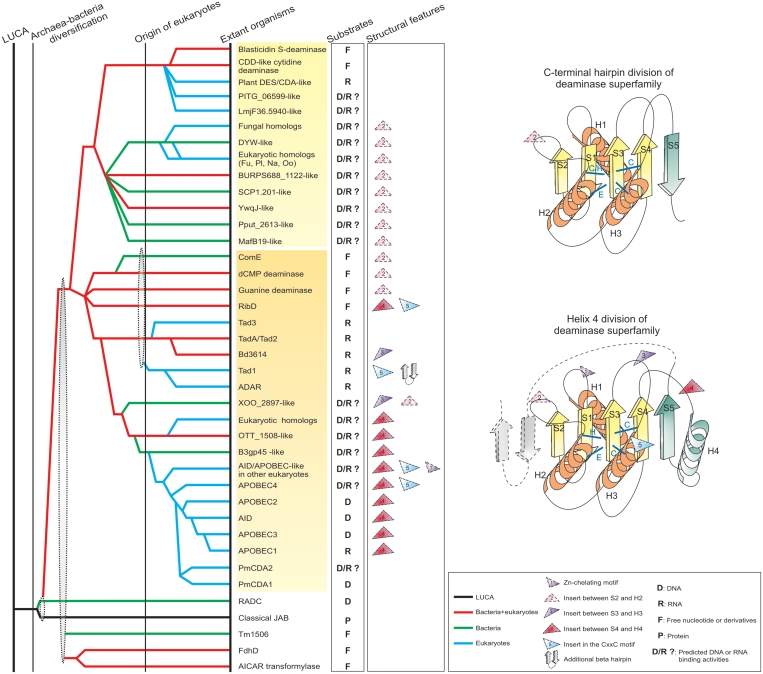

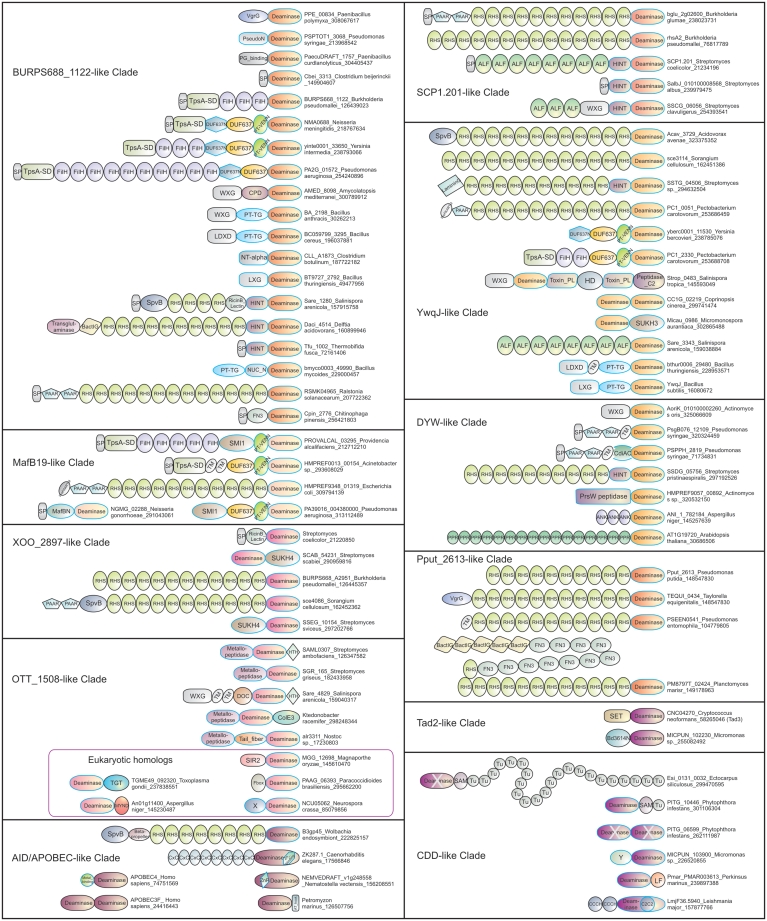

References
-
- Harold CS. RNA and DNA Editing: Molecular Mechanisms and Their Integration into Biological Systems. Hoboken, New Jersey: John Wiley & Sons, Inc; 2008.
-
- Kumasaka T, Yamamoto M, Furuichi M, Nakasako M, Teh AH, Kimura M, Yamaguchi I, Ueki T. Crystal structures of blasticidin S deaminase (BSD): implications for dynamic properties of catalytic zinc. J. Biol. Chem. 2007;282:37103–37111. - PubMed
-
- Stenmark P, Moche M, Gurmu D, Nordlund P. The crystal structure of the bifunctional deaminase/reductase RibD of the riboflavin biosynthetic pathway in Escherichia coli: implications for the reductive mechanism. J. Mol. Biol. 2007;373:48–64. - PubMed
-
- Hamilton CE, Papavasiliou FN, Rosenberg BR. Diverse functions for DNA and RNA editing in the immune system. RNA Biol. 2010;7:220–228. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
