

Toxoplasma gondii effectors are master regulators of the inflammatory response
- PMID: 21893432
- PMCID: PMC3200456
- DOI: 10.1016/j.pt.2011.08.001
Toxoplasma gondii effectors are master regulators of the inflammatory response
Abstract
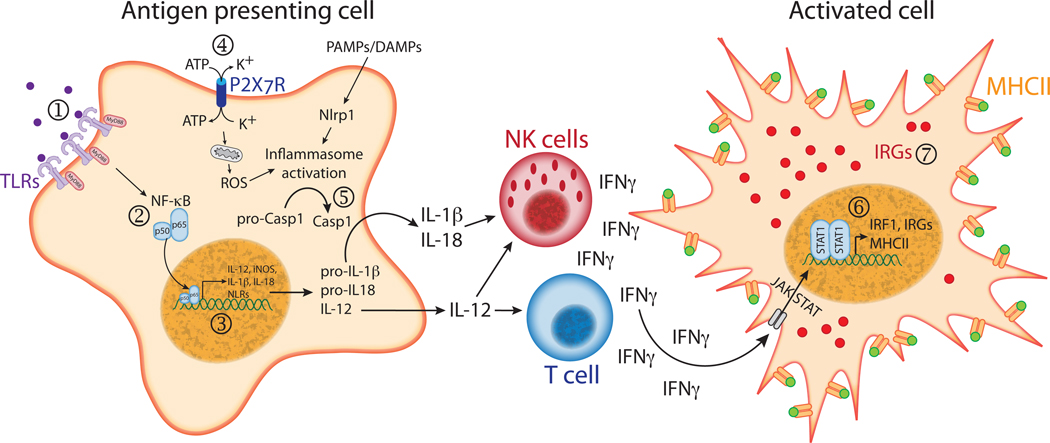
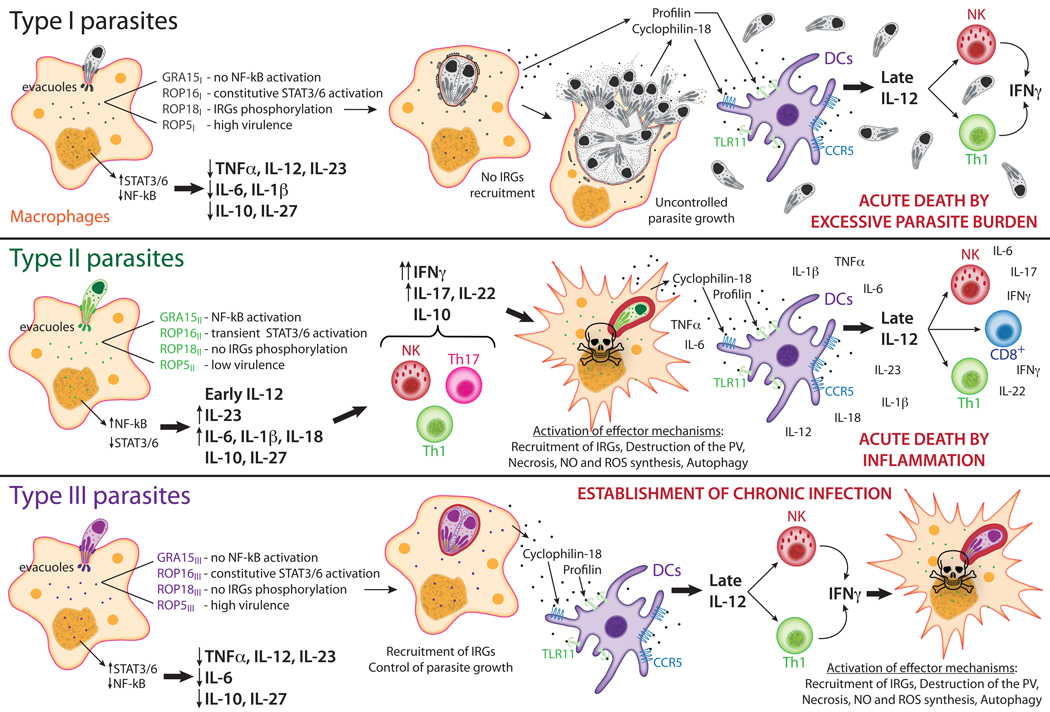
Toxoplasma is a highly successful parasite that establishes a life-long chronic infection. To do this, it must carefully regulate immune activation and host cell effector mechanisms. Here we review the latest developments in our understanding of how Toxoplasma counteracts the immune response of the host, and in some cases provokes it, through the use of specific parasite effector proteins. An emerging theme from these discoveries is that Toxoplasma effectors are master regulators of the pro-inflammatory response, which elicits many of the toxoplasmacidal mechanisms of the host. We speculate that combinations of these effectors present in certain Toxoplasma strains work to maintain an optimal parasite burden in different hosts to ensure parasite transmission.
Copyright © 2011 Elsevier Ltd. All rights reserved.
Figures
References
-
- Dubremetz JF. Host cell invasion by Toxoplasma gondii. Trends Microbiol. 1998;6:27–30. - PubMed
-
- Hill D, Dubey JP. Toxoplasma gondii: transmission, diagnosis and prevention. Clin Microbiol Infect. 2002;8:634–640. - PubMed
-
- Morisaki JH, et al. Invasion of Toxoplasma gondii occurs by active penetration of the host cell. Journal of Cell Science. 1995;108(Pt 6):2457–2464. - PubMed
-
- Plattner F, Soldati-Favre D. Hijacking of host cellular functions by the Apicomplexa. Annu Rev Microbiol. 2008;62:471–487. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
