

Detection of classical 17p11.2 deletions, an atypical deletion and RAI1 alterations in patients with features suggestive of Smith-Magenis syndrome
- PMID: 21897445
- PMCID: PMC3260931
- DOI: 10.1038/ejhg.2011.167
Detection of classical 17p11.2 deletions, an atypical deletion and RAI1 alterations in patients with features suggestive of Smith-Magenis syndrome
Abstract
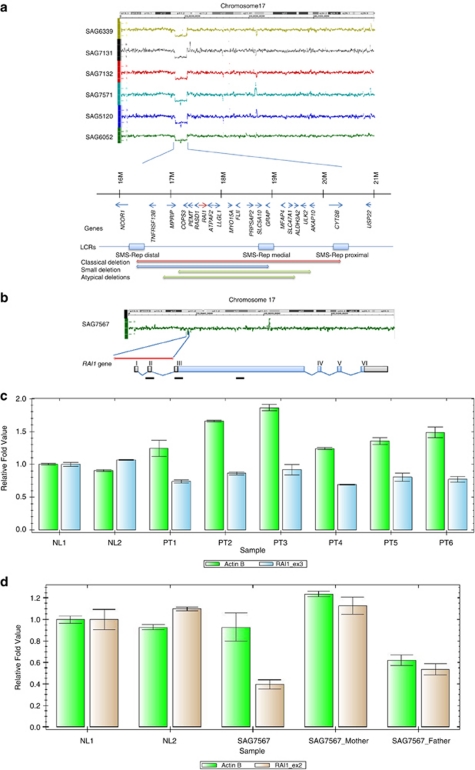
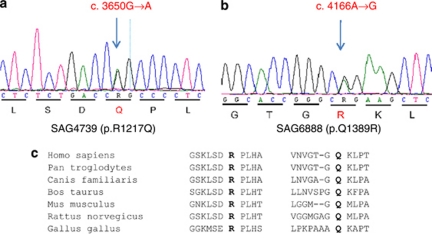
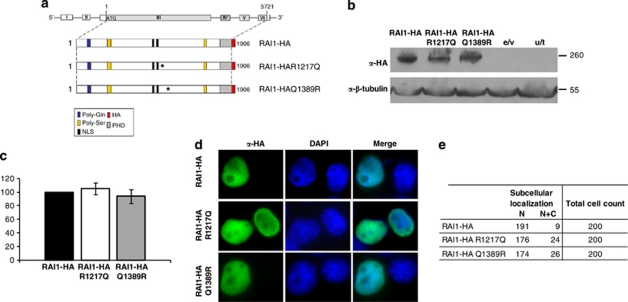
Smith-Magenis syndrome (SMS) is a complex disorder whose clinical features include mild to severe intellectual disability with speech delay, growth failure, brachycephaly, flat midface, short broad hands, and behavioral problems. SMS is typically caused by a large deletion on 17p11.2 that encompasses multiple genes including the retinoic acid induced 1, RAI1, gene or a mutation in the RAI1 gene. Here we have evaluated 30 patients with suspected SMS and identified SMS-associated classical 17p11.2 deletions in six patients, an atypical deletion of ~139 kb that partially deletes the RAI1 gene in one patient, and RAI1 gene nonsynonymous alterations of unknown significance in two unrelated patients. The RAI1 mutant proteins showed no significant alterations in molecular weight, subcellular localization and transcriptional activity. Clinical features of patients with or without 17p11.2 deletions and mutations involving the RAI1 gene were compared to identify phenotypes that may be useful in diagnosing patients with SMS.
Figures

Similar articles
-
Molecular analysis of the Retinoic Acid Induced 1 gene (RAI1) in patients with suspected Smith-Magenis syndrome without the 17p11.2 deletion.PLoS One. 2011;6(8):e22861. doi: 10.1371/journal.pone.0022861. Epub 2011 Aug 8. PLoS One. 2011. PMID: 21857958 Free PMC article.
-
Delayed diagnosis in a house of correction: Smith-Magenis syndrome due to a de novo nonsense RAI1 variant.Am J Med Genet A. 2016 Sep;170(9):2383-8. doi: 10.1002/ajmg.a.37602. Epub 2016 Jun 17. Am J Med Genet A. 2016. PMID: 27311559 Free PMC article.
-
Genotype-phenotype correlation in Smith-Magenis syndrome: evidence that multiple genes in 17p11.2 contribute to the clinical spectrum.Genet Med. 2006 Jul;8(7):417-27. doi: 10.1097/01.gim.0000228215.32110.89. Genet Med. 2006. PMID: 16845274
-
Smith-Magenis Syndrome-Clinical Review, Biological Background and Related Disorders.Genes (Basel). 2022 Feb 11;13(2):335. doi: 10.3390/genes13020335. Genes (Basel). 2022. PMID: 35205380 Free PMC article. Review.
-
Intellectual and Behavioral Phenotypes of Smith-Magenis Syndrome: Comparisons between Individuals with a 17p11.2 Deletion and Pathogenic RAI1 Variant.Genes (Basel). 2023 Jul 25;14(8):1514. doi: 10.3390/genes14081514. Genes (Basel). 2023. PMID: 37628566 Free PMC article. Review.
Cited by
-
A unique Smith-Magenis patient with a de novo intragenic deletion on the maternally inherited overexpressed RAI1 allele.Eur J Hum Genet. 2022 Nov;30(11):1233-1238. doi: 10.1038/s41431-022-01143-5. Epub 2022 Jul 11. Eur J Hum Genet. 2022. PMID: 35821519 Free PMC article.
-
Identification of Nine New RAI1-Truncating Mutations in Smith-Magenis Syndrome Patients without 17p11.2 Deletions.Mol Syndromol. 2014 Feb;5(2):57-64. doi: 10.1159/000357359. Epub 2014 Jan 7. Mol Syndromol. 2014. PMID: 24715852 Free PMC article.
-
A case of Smith-Magenis syndrome with skin manifestations caused by a novel locus mutation in the RAI1 gene.J Int Med Res. 2023 Sep;51(9):3000605231190553. doi: 10.1177/03000605231190553. J Int Med Res. 2023. PMID: 37756600 Free PMC article.
-
Rai1 Haploinsufficiency Is Associated with Social Abnormalities in Mice.Biology (Basel). 2017 Apr 27;6(2):25. doi: 10.3390/biology6020025. Biology (Basel). 2017. PMID: 28448442 Free PMC article.
-
Whole Exome Sequencing Reveals Homozygous Mutations in RAI1, OTOF, and SLC26A4 Genes Associated with Nonsyndromic Hearing Loss in Altaian Families (South Siberia).PLoS One. 2016 Apr 15;11(4):e0153841. doi: 10.1371/journal.pone.0153841. eCollection 2016. PLoS One. 2016. PMID: 27082237 Free PMC article.
References
-
- Smith ACM, Magenis RE, Elsea SH. Overview of Smith-Magenis syndrome. J Assoc Genet Technol. 2005;31:163–167. - PubMed
-
- Elsea SH, Girirajan S. Smith-Magenis syndrome. Eur J Hum Genet. 2008;16:412–421. - PubMed
-
- Smith ACM, Boyd K, Elsea SH, et al. GeneReviews at GeneTests: Medical Genetics Information Resource (database online) Seattle: Copyright, University of Washington; 1997–2010. Smith-Magenis Syndrome.
-
- Dykens EM, Finucane BM, Gayley C. Brief report: cognitive and behavioral profiles in persons with Smith-Magenis syndrome. J Autism Dev Disord. 1997;27:203–211. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
