

Genetic interactions between hepatocyte nuclear factor-6 and Notch signaling regulate mouse intrahepatic bile duct development in vivo
- PMID: 21898486
- PMCID: PMC3235248
- DOI: 10.1002/hep.24631
Genetic interactions between hepatocyte nuclear factor-6 and Notch signaling regulate mouse intrahepatic bile duct development in vivo
Abstract
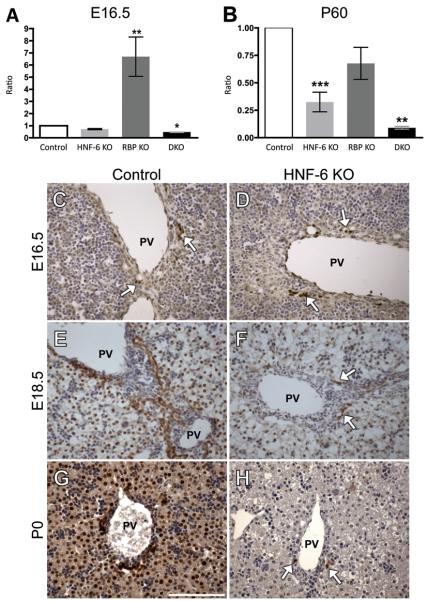
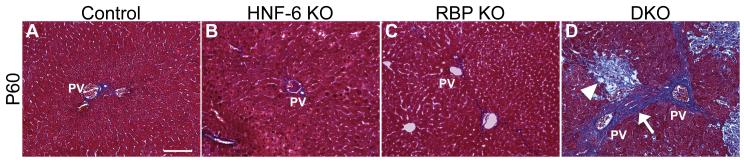
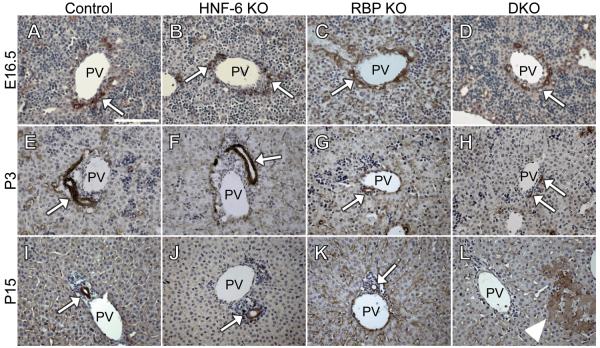
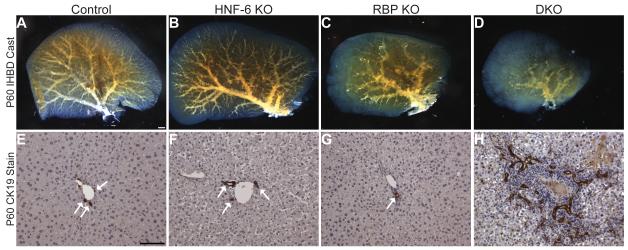
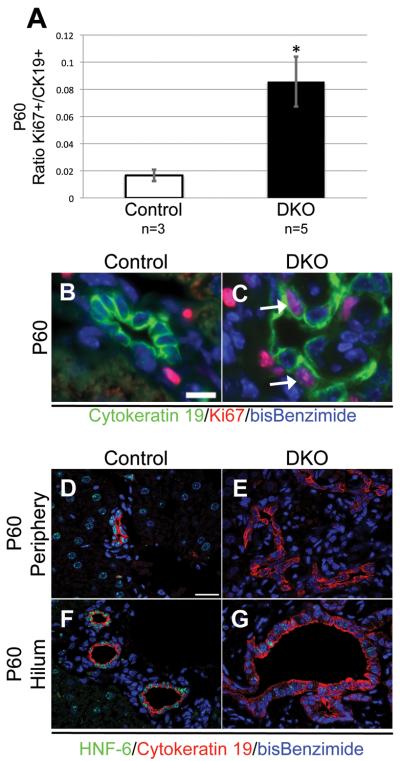
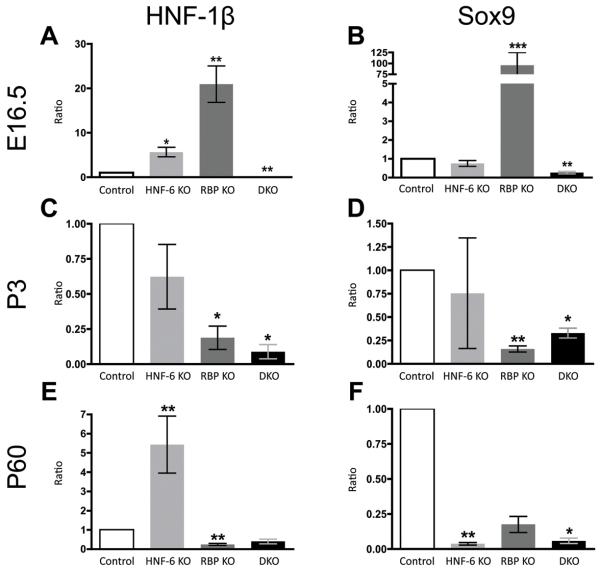
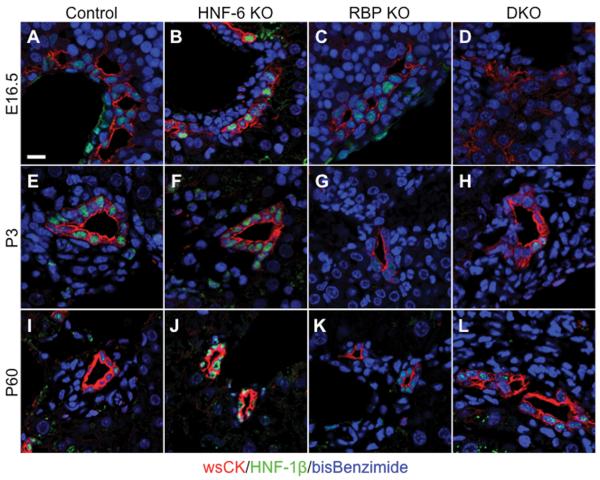
Notch signaling and hepatocyte nuclear factor-6 (HNF-6) are two genetic factors known to affect lineage commitment in the bipotential hepatoblast progenitor cell (BHPC) population. A genetic interaction involving Notch signaling and HNF-6 in mice has been inferred through separate experiments showing that both affect BHPC specification and bile duct morphogenesis. To define the genetic interaction between HNF-6 and Notch signaling in an in vivo mouse model, we examined the effects of BHPC-specific loss of HNF-6 alone and within the background of BHPC-specific loss of recombination signal binding protein immunoglobulin kappa J (RBP-J), the common DNA-binding partner of all Notch receptors. Isolated loss of HNF-6 in this mouse model fails to demonstrate a phenotypic variance in bile duct development compared to control. However, when HNF-6 loss is combined with RBP-J loss, a phenotype consisting of cholestasis, hepatic necrosis, and fibrosis is observed that is more severe than the phenotype seen with Notch signaling loss alone. This phenotype is associated with significant intrahepatic biliary system abnormalities, including an early decrease in biliary epithelial cells, evolving to ductular proliferation and a decrease in the density of communicating peripheral bile duct branches. In this in vivo model, simultaneous loss of both HNF-6 and RBP-J results in down-regulation of both HNF-1β and Sox9 (sex determining region Y-related HMG box transcription factor 9).
Conclusion: HNF-6 and Notch signaling interact in vivo to control expression of downstream mediators essential to the normal development of the intrahepatic biliary system. This study provides a model to investigate genetic interactions of factors important to intrahepatic bile duct development and their effect on cholestatic liver disease phenotypes.
Copyright © 2011 American Association for the Study of Liver Diseases.
Figures
References
-
- Chiba S. Notch signaling in stem cell systems. Stem Cells. 2006;24:2437–2447. - PubMed
-
- Li L, Krantz ID, Deng Y, Genin A, Banta AB, Collins CC, Qi M, et al. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat Genet. 1997;16:243–251. - PubMed
-
- Oda T, Elkahloun AG, Pike BL, Okajima K, Krantz ID, Genin A, Piccoli DA, et al. Mutations in the human Jagged1 gene are responsible for Alagille syndrome. Nat Genet. 1997;16:235–242. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials