

Chemical and biological approaches for adapting proteostasis to ameliorate protein misfolding and aggregation diseases: progress and prognosis
- PMID: 21900404
- PMCID: PMC3225948
- DOI: 10.1101/cshperspect.a004507
Chemical and biological approaches for adapting proteostasis to ameliorate protein misfolding and aggregation diseases: progress and prognosis
Abstract
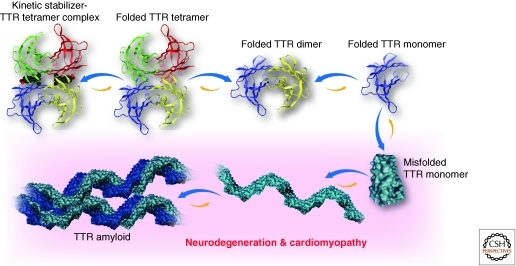
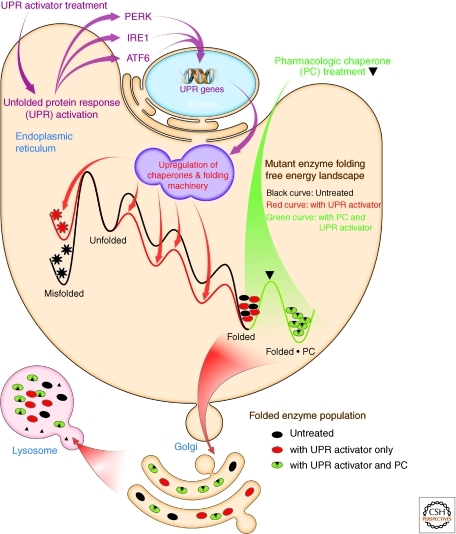
Maintaining the proteome to preserve the health of an organism in the face of developmental changes, environmental insults, infectious diseases, and rigors of aging is a formidable task. The challenge is magnified by the inheritance of mutations that render individual proteins subject to misfolding and/or aggregation. Maintenance of the proteome requires the orchestration of protein synthesis, folding, degradation, and trafficking by highly conserved/deeply integrated cellular networks. In humans, no less than 2000 genes are involved. Stress sensors detect the misfolding and aggregation of proteins in specific organelles and respond by activating stress-responsive signaling pathways. These culminate in transcriptional and posttranscriptional programs that up-regulate the homeostatic mechanisms unique to that organelle. Proteostasis is also strongly influenced by the general properties of protein folding that are intrinsic to every proteome. These include the kinetics and thermodynamics of the folding, misfolding, and aggregation of individual proteins. We examine a growing body of evidence establishing that when cellular proteostasis goes awry, it can be reestablished by deliberate chemical and biological interventions. We start with approaches that employ chemicals or biological agents to enhance the general capacity of the proteostasis network. We then introduce chemical approaches to prevent the misfolding or aggregation of specific proteins through direct binding interactions. We finish with evidence that synergy is achieved with the combination of mechanistically distinct approaches to reestablish organismal proteostasis.
Figures
Similar articles
-
Underlying mechanisms and chemical/biochemical therapeutic approaches to ameliorate protein misfolding neurodegenerative diseases.Biofactors. 2017 Nov;43(6):737-759. doi: 10.1002/biof.1264. Epub 2016 Feb 22. Biofactors. 2017. PMID: 26899445 Review.
-
Pharmacologic Approaches for Adapting Proteostasis in the Secretory Pathway to Ameliorate Protein Conformational Diseases.Cold Spring Harb Perspect Biol. 2020 May 1;12(5):a034108. doi: 10.1101/cshperspect.a034108. Cold Spring Harb Perspect Biol. 2020. PMID: 31088828 Free PMC article. Review.
-
Emergent properties of proteostasis in managing cystic fibrosis.Cold Spring Harb Perspect Biol. 2011 Feb 1;3(2):a004499. doi: 10.1101/cshperspect.a004499. Cold Spring Harb Perspect Biol. 2011. PMID: 21421917 Free PMC article. Review.
-
Proteostasis: bad news and good news from the endoplasmic reticulum.Swiss Med Wkly. 2014 Aug 21;144:w14001. doi: 10.4414/smw.2014.14001. eCollection 2014. Swiss Med Wkly. 2014. PMID: 25144910 Review.
-
Organismal Protein Homeostasis Mechanisms.Genetics. 2020 Aug;215(4):889-901. doi: 10.1534/genetics.120.301283. Genetics. 2020. PMID: 32759342 Free PMC article. Review.
Cited by
-
AAA+ Protein-Based Technologies to Counter Neurodegenerative Disease.Biophys J. 2019 Apr 23;116(8):1380-1385. doi: 10.1016/j.bpj.2019.03.007. Epub 2019 Mar 22. Biophys J. 2019. PMID: 30952364 Free PMC article. Review.
-
Regulating Secretory Proteostasis through the Unfolded Protein Response: From Function to Therapy.Trends Cell Biol. 2017 Oct;27(10):722-737. doi: 10.1016/j.tcb.2017.05.006. Epub 2017 Jun 21. Trends Cell Biol. 2017. PMID: 28647092 Free PMC article. Review.
-
Endoplasmic reticulum quality control and systemic amyloid disease: Impacting protein stability from the inside out.IUBMB Life. 2015 Jun;67(6):404-13. doi: 10.1002/iub.1386. Epub 2015 May 26. IUBMB Life. 2015. PMID: 26018985 Free PMC article. Review.
-
Engineering enhanced protein disaggregases for neurodegenerative disease.Prion. 2015;9(2):90-109. doi: 10.1080/19336896.2015.1020277. Prion. 2015. PMID: 25738979 Free PMC article. Review.
-
Protein homeostasis as a therapeutic target for diseases of protein conformation.Curr Top Med Chem. 2012;12(22):2623-40. doi: 10.2174/1568026611212220014. Curr Top Med Chem. 2012. PMID: 23339312 Free PMC article. Review.
References
-
- Adamski-Werner SL, Palaninathan SK, Sacchettini JC, Kelly JW 2004. Diflunisal analogues stabilize the native state of transthyretin. Potent inhibition of amyloidogenesis. J Med Chem 47: 355–374 - PubMed
-
- Alfonso P, Pampin S, Estrada J, Rodriguez-Rey JC, Giraldo P, Sancho J, Pocovi M 2005. Miglustat (NB-DNJ) works as a chaperone for mutated acid β-glucosidase in cells transfected with several Gaucher disease mutations. Blood Cells Molecules Diseases 35: 268–276 - PubMed
-
- Andrade C 1952. A peculiar form of peripheral neuropathy; Familiar atypical generalized amyloidosis with special involvement of the peripheral nerves. Brain 75: 408–427 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources