

Selective hippocampal neurodegeneration in transgenic mice expressing small amounts of truncated Aβ is induced by pyroglutamate-Aβ formation
- PMID: 21900558
- PMCID: PMC6623394
- DOI: 10.1523/JNEUROSCI.1794-11.2011
Selective hippocampal neurodegeneration in transgenic mice expressing small amounts of truncated Aβ is induced by pyroglutamate-Aβ formation
Abstract
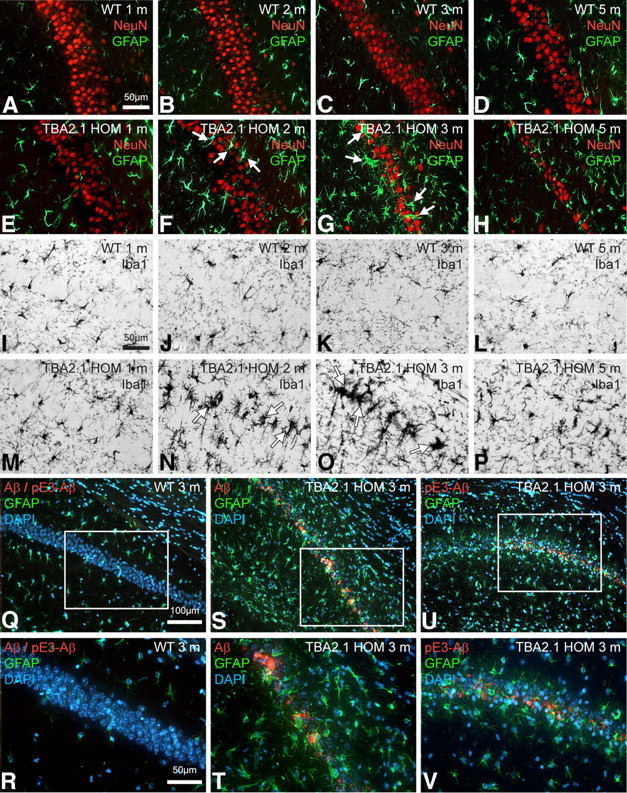
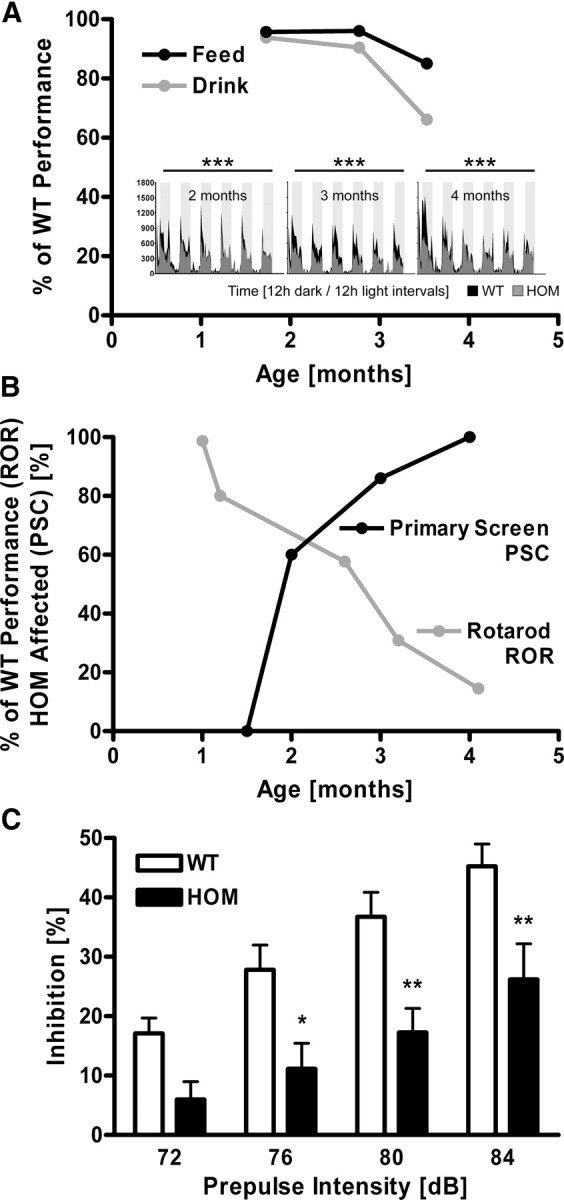
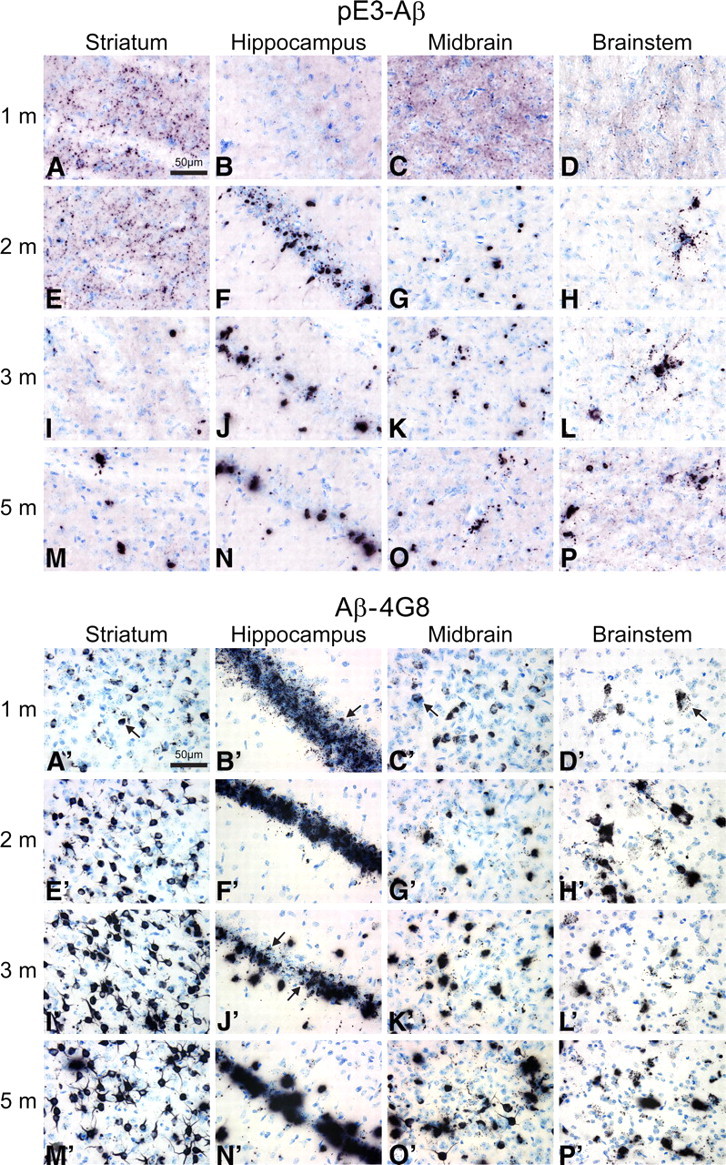
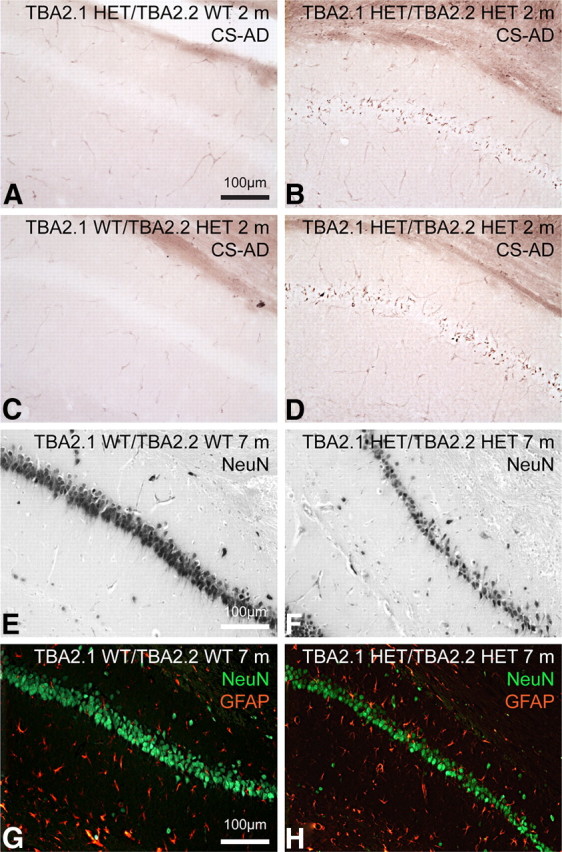
Posttranslational amyloid-β (Aβ) modification is considered to play an important role in Alzheimer's disease (AD) etiology. An N-terminally modified Aβ species, pyroglutamate-amyloid-β (pE3-Aβ), has been described as a major constituent of Aβ deposits specific to human AD but absent in normal aging. Formed via cyclization of truncated Aβ species by glutaminyl cyclase (QC; QPCT) and/or its isoenzyme (isoQC; QPCTL), pE3-Aβ aggregates rapidly and is known to seed additional Aβ aggregation. To directly investigate pE3-Aβ toxicity in vivo, we generated and characterized transgenic TBA2.1 and TBA2.2 mice, which express truncated mutant human Aβ. Along with a rapidly developing behavioral phenotype, these mice showed progressively accumulating Aβ and pE3-Aβ deposits in brain regions of neuronal loss, impaired long-term potentiation, microglial activation, and astrocytosis. Illustrating a threshold for pE3-Aβ neurotoxicity, this phenotype was not found in heterozygous animals but in homozygous TBA2.1 or double-heterozygous TBA2.1/2.2 animals only. A significant amount of pE3-Aβ formation was shown to be QC-dependent, because crossbreeding of TBA2.1 with QC knock-out, but not isoQC knock-out, mice significantly reduced pE3-Aβ levels. Hence, lowering the rate of QC-dependent posttranslational pE3-Aβ formation can, in turn, lower the amount of neurotoxic Aβ species in AD.
Figures




References
-
- Augustin M, Sedlmeier R, Peters T, Huffstadt U, Kochmann E, Simon D, Schöniger M, Garke-Mayerthaler S, Laufs J, Mayhaus M, Franke S, Klose M, Graupner A, Kurzmann M, Zinser C, Wolf A, Voelkel M, Kellner M, Kilian M, Seelig S, Koppius A, Teubner A, Korthaus D, Nehls M, Wattler S. Efficient and fast targeted production of murine models based on ENU mutagenesis. Mamm Genome. 2005;16:405–413. - PubMed
-
- Bayer TA, Wirths O. Review on the APP/PS1KI mouse model: intraneuronal Abeta accumulation triggers axonopathy, neuron loss and working memory impairment. Genes Brain Behav. 2008;7(Suppl 1):6–11. - PubMed
-
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous