

Evolution at the subgene level: domain rearrangements in the Drosophila phylogeny
- PMID: 21900599
- PMCID: PMC3258039
- DOI: 10.1093/molbev/msr222
Evolution at the subgene level: domain rearrangements in the Drosophila phylogeny
Abstract
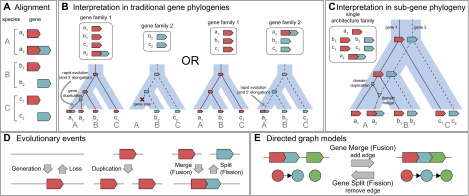
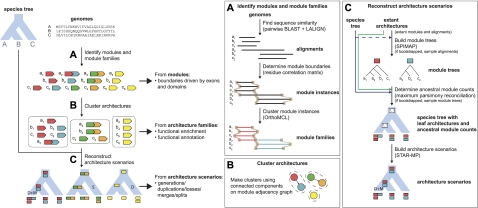
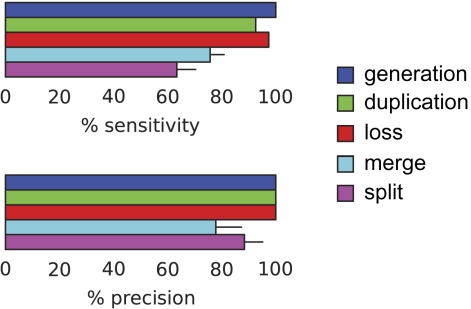
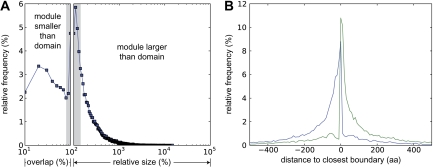
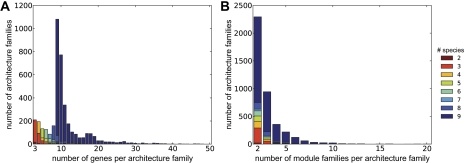
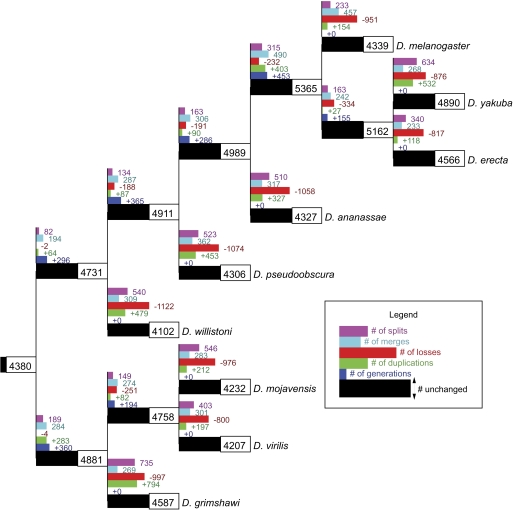
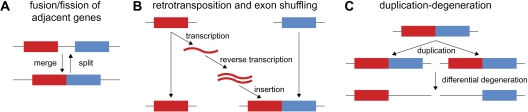
Although the possibility of gene evolution by domain rearrangements has long been appreciated, current methods for reconstructing and systematically analyzing gene family evolution are limited to events such as duplication, loss, and sometimes, horizontal transfer. However, within the Drosophila clade, we find domain rearrangements occur in 35.9% of gene families, and thus, any comprehensive study of gene evolution in these species will need to account for such events. Here, we present a new computational model and algorithm for reconstructing gene evolution at the domain level. We develop a method for detecting homologous domains between genes and present a phylogenetic algorithm for reconstructing maximum parsimony evolutionary histories that include domain generation, duplication, loss, merge (fusion), and split (fission) events. Using this method, we find that genes involved in fusion and fission are enriched in signaling and development, suggesting that domain rearrangements and reuse may be crucial in these processes. We also find that fusion is more abundant than fission, and that fusion and fission events occur predominantly alongside duplication, with 92.5% and 34.3% of fusion and fission events retaining ancestral architectures in the duplicated copies. We provide a catalog of ∼9,000 genes that undergo domain rearrangement across nine sequenced species, along with possible mechanisms for their formation. These results dramatically expand on evolution at the subgene level and offer several insights into how new genes and functions arise between species.
Figures
References
-
- Apic G, Gough J, Teichmann SA. An insight into domain combinations. Bioinformatics. 2001;17:S83–S89. - PubMed
-
- Apic G, Huber W, Teichmann SA. Multi-domain protein families and domain pairs: comparison with known structures and a random model of domain recombination. J Struct Funct Genomics. 2003;4:67–78. - PubMed
-
- Arvestad L, Lagergren J, Sennblad B. The gene evolution model and computing its associated probabilities. JACM. 2009;56:1–44.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
