

The Role of BH3-Only Proteins in Tumor Cell Development, Signaling, and Treatment
- PMID: 21901166
- PMCID: PMC3161420
- DOI: 10.1177/1947601911417177
The Role of BH3-Only Proteins in Tumor Cell Development, Signaling, and Treatment
Abstract
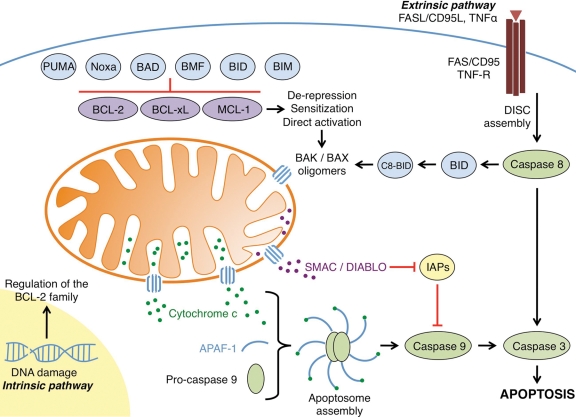
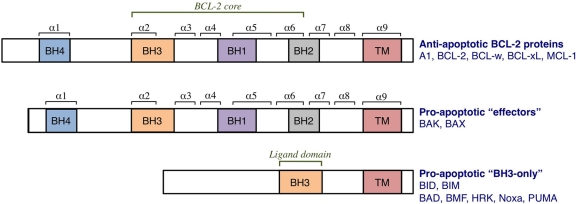
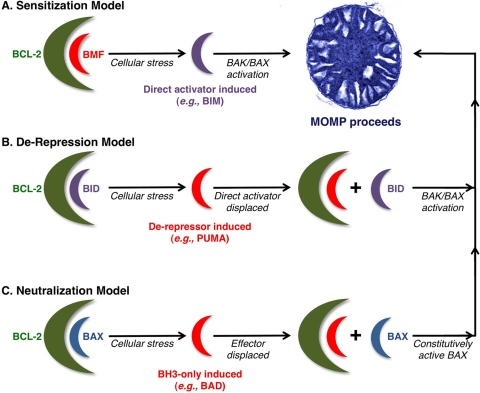
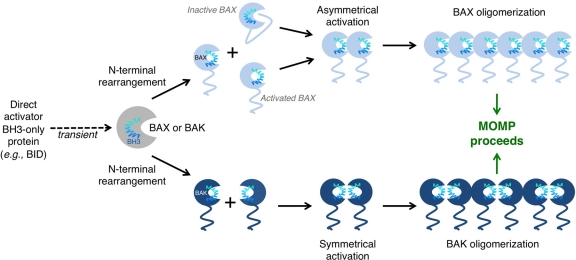
Tumor cells have devised several strategies to block the mitochondrial pathway of apoptosis despite endogenous or pharmacological cues to die. This process of cell death proceeds through the coordinated regulation of multiple anti-apoptotic and pro-apoptotic BCL-2 family proteins that ultimately impinge on the integrity of the outer mitochondrial membrane. Once compromised, mitochondria release pro-apoptotic factors to promote caspase activation and the apoptotic phenotype. Within the BCL-2 family exists a subclass of pro-apoptotic members termed the BH3-only proteins, which directly and/or indirectly functionally regulate the remaining anti- and pro-apoptotic BCL-2 proteins to compromise mitochondria and engage apoptosis. The focus of this review is to discuss the cellular and pharmacological regulation of the BH3-only proteins to gain a better understanding of the signaling pathways and agents that regulate this class of proteins. As the BH3-only proteins increase cellular sensitivity to pro-apoptotic agents such as chemotherapeutics, numerous small-molecule BH3 mimetics have been developed and are currently in various phases of clinical trials. Toward the end of the review, the discovery and application of the small-molecule BH3 mimetics will be discussed.
Keywords: BCL-2 family; BH3-only proteins; apoptosis; cancer; mitochondria.
Conflict of interest statement
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Figures
References
-
- Barr PJ, Tomei LD. Apoptosis and its role in human disease. Biotechnology (N Y). 1994;12:487-93 - PubMed
-
- McDonnell TJ, Korsmeyer SJ. Progression from lymphoid hyperplasia to high-grade malignant lymphoma in mice transgenic for the t(14; 18). Nature. 1991;349:254-6 - PubMed
-
- Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456-62 - PubMed
-
- Watanabe-Fukunaga R, Brannan CI, Copeland NG, Jenkins NA, Nagata S. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature. 1992;356:314-7 - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials