

Genetic diagnosis of X-linked dominant Hypophosphatemic Rickets in a cohort study: tubular reabsorption of phosphate and 1,25(OH)2D serum levels are associated with PHEX mutation type
- PMID: 21902834
- PMCID: PMC3189111
- DOI: 10.1186/1471-2350-12-116
Genetic diagnosis of X-linked dominant Hypophosphatemic Rickets in a cohort study: tubular reabsorption of phosphate and 1,25(OH)2D serum levels are associated with PHEX mutation type
Abstract
Background: Genetic Hypophosphatemic Rickets (HR) is a group of diseases characterized by renal phosphate wasting with inappropriately low or normal 1,25-dihydroxyvitamin D3 (1,25(OH)2D) serum levels. The most common form of HR is X-linked dominant HR (XLHR) which is caused by inactivating mutations in the PHEX gene. The purpose of this study was to perform genetic diagnosis in a cohort of patients with clinical diagnosis of HR, to perform genotype-phenotype correlations of those patients and to compare our data with other HR cohort studies.
Methods: Forty three affected individuals from 36 non related families were analyzed. For the genetic analysis, the PHEX gene was sequenced in all of the patients and in 13 cases the study was complemented by mRNA sequencing and Multiple Ligation Probe Assay. For the genotype-phenotype correlation study, the clinical and biochemical phenotype of the patients was compared with the type of mutation, which was grouped into clearly deleterious or likely causative, using the Mann-Whitney and Fisher's exact test.
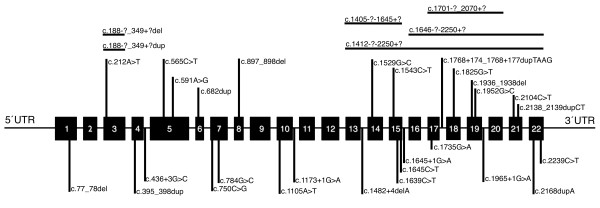
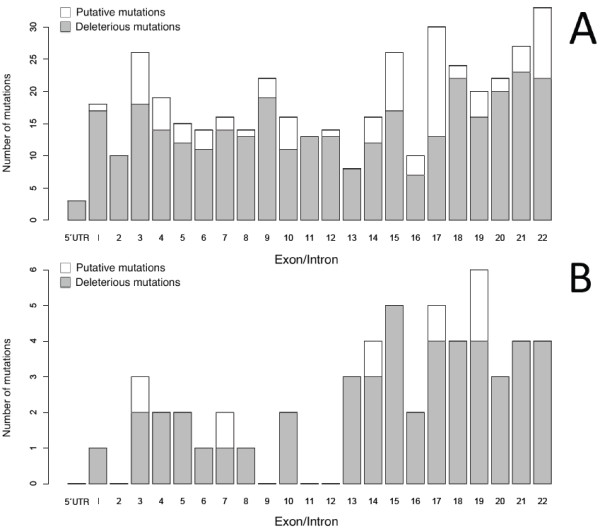
Results: Mutations in the PHEX gene were identified in all the patients thus confirming an XLHR. Thirty four different mutations were found distributed throughout the gene with higher density at the 3' end. The majority of the mutations were novel (69.4%), most of them resulted in a truncated PHEX protein (83.3%) and were family specific (88.9%). Tubular reabsorption of phosphate (TRP) and 1,25(OH)2D serum levels were significantly lower in patients carrying clearly deleterious mutations than in patients carrying likely causative ones (61.39 ± 19.76 vs. 80.14 ± 8.80%, p = 0.028 and 40.93 ± 30.73 vs. 78.46 ± 36.27 pg/ml, p = 0.013).
Conclusions: PHEX gene mutations were found in all the HR cases analyzed, which was in contrast with other cohort studies. Patients with clearly deleterious PHEX mutations had lower TRP and 1,25(OH)2D levels suggesting that the PHEX type of mutation might predict the XLHR phenotype severity.
Figures

References
-
- Lorenz-Depiereux B, Bastepe M, Benet-Pages A, Amyere M, Wagenstaller J, Muller-Barth U, Badenhoop K, Kaiser SM, Rittmaster RS, Shlossberg AH, Olivares JL, Loris C, Ramos FJ, Glorieux F, Vikkula M, Jüppner H, Strom TM. et al. DMP1 mutations in autosomal recessive hypophosphatemia implicate a bone matrix protein in the regulation of phosphate homeostasis. Nat Genet. 2006;38(11):1248–1250. doi: 10.1038/ng1868. - DOI - PMC - PubMed
