

G protein-coupled receptor kinases: more than just kinases and not only for GPCRs
- PMID: 21903131
- PMCID: PMC3241883
- DOI: 10.1016/j.pharmthera.2011.08.001
G protein-coupled receptor kinases: more than just kinases and not only for GPCRs
Abstract
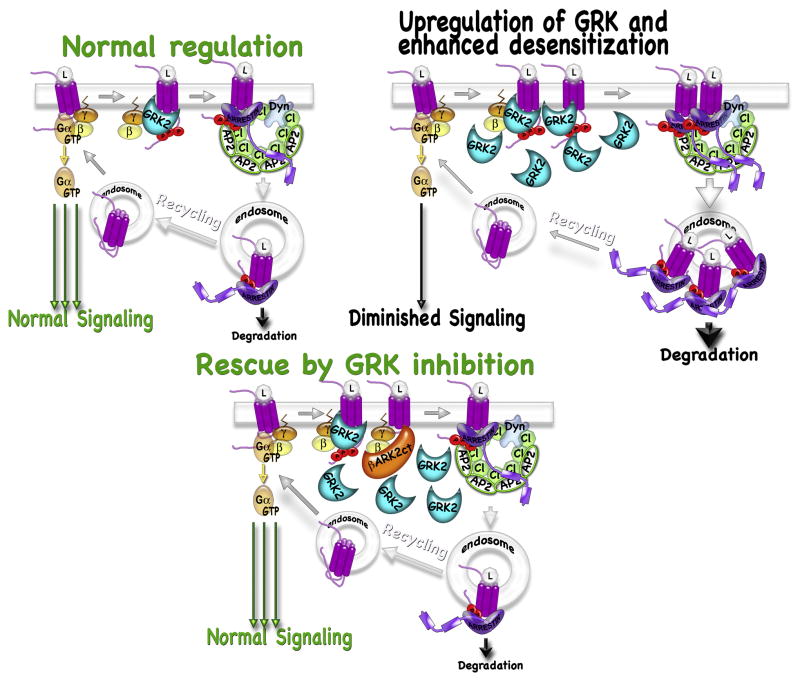
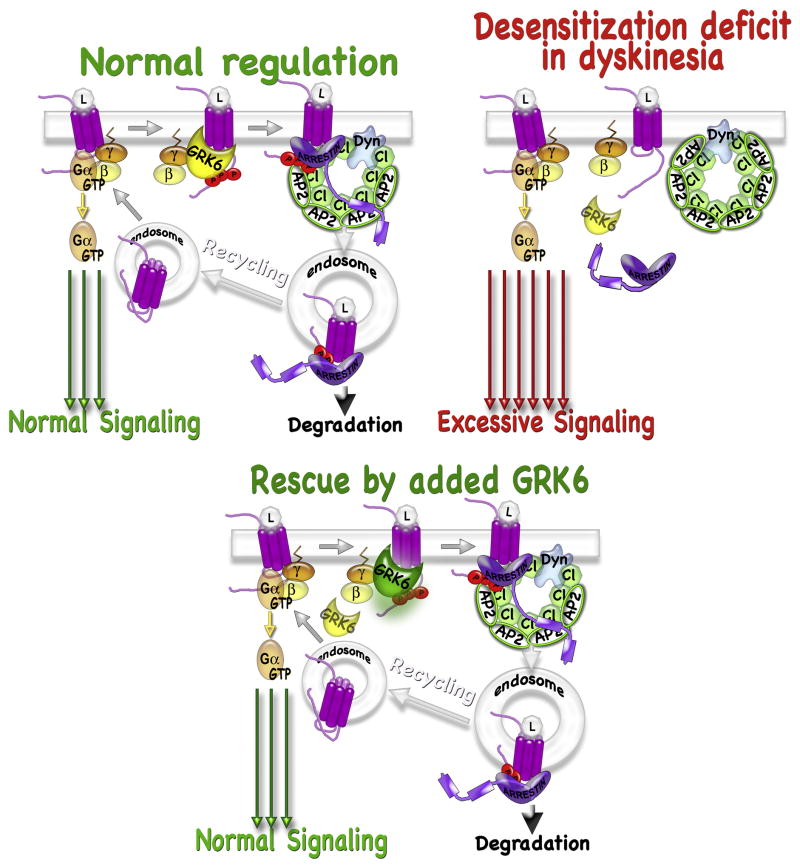
G protein-coupled receptor (GPCR) kinases (GRKs) are best known for their role in homologous desensitization of GPCRs. GRKs phosphorylate activated receptors and promote high affinity binding of arrestins, which precludes G protein coupling. GRKs have a multidomain structure, with the kinase domain inserted into a loop of a regulator of G protein signaling homology domain. Unlike many other kinases, GRKs do not need to be phosphorylated in their activation loop to achieve an activated state. Instead, they are directly activated by docking with active GPCRs. In this manner they are able to selectively phosphorylate Ser/Thr residues on only the activated form of the receptor, unlike related kinases such as protein kinase A. GRKs also phosphorylate a variety of non-GPCR substrates and regulate several signaling pathways via direct interactions with other proteins in a phosphorylation-independent manner. Multiple GRK subtypes are present in virtually every animal cell, with the highest expression levels found in neurons, with their extensive and complex signal regulation. Insufficient or excessive GRK activity was implicated in a variety of human disorders, ranging from heart failure to depression to Parkinson's disease. As key regulators of GPCR-dependent and -independent signaling pathways, GRKs are emerging drug targets and promising molecular tools for therapy. Targeted modulation of expression and/or of activity of several GRK isoforms for therapeutic purposes was recently validated in cardiac disorders and Parkinson's disease.
Copyright © 2011 Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors declare that there are no conflicts of interests.
Figures

References
-
- Alloway PG, Howard L, Dolph PJ. The formation of stable rhodopsin-arrestin complexes induces apoptosis and photoreceptor cell degeneration. Neuron. 2000;28:129–138. - PubMed
-
- Ambrose C, James M, Barnes G, Lin C, Bates G, Altherr M, et al. A novel G protein-coupled receptor kinase gene cloned from 4p16.3. Hum Mol Genet. 1992;1:697–703. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
