

A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data
- PMID: 21903627
- PMCID: PMC3198575
- DOI: 10.1093/bioinformatics/btr509
A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data
Abstract
Motivation: Most existing methods for DNA sequence analysis rely on accurate sequences or genotypes. However, in applications of the next-generation sequencing (NGS), accurate genotypes may not be easily obtained (e.g. multi-sample low-coverage sequencing or somatic mutation discovery). These applications press for the development of new methods for analyzing sequence data with uncertainty.
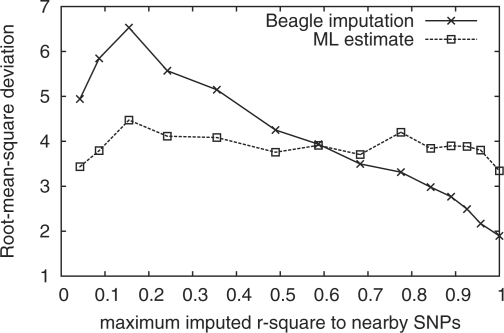
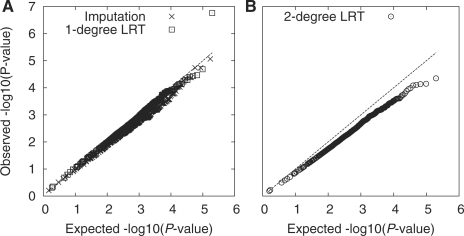
Results: We present a statistical framework for calling SNPs, discovering somatic mutations, inferring population genetical parameters and performing association tests directly based on sequencing data without explicit genotyping or linkage-based imputation. On real data, we demonstrate that our method achieves comparable accuracy to alternative methods for estimating site allele count, for inferring allele frequency spectrum and for association mapping. We also highlight the necessity of using symmetric datasets for finding somatic mutations and confirm that for discovering rare events, mismapping is frequently the leading source of errors.
Availability: http://samtools.sourceforge.net.
Contact: hengli@broadinstitute.org.
Figures

References
-
- Brent RP. Algorithms for Minimization without Derivatives. Englewood Cliffs, New Jersey: Prentice-Hall; 1973.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
