

An autosomal analysis gives no genetic evidence for complex speciation of humans and chimpanzees
- PMID: 21903679
- PMCID: PMC3299331
- DOI: 10.1093/molbev/msr172
An autosomal analysis gives no genetic evidence for complex speciation of humans and chimpanzees
Abstract
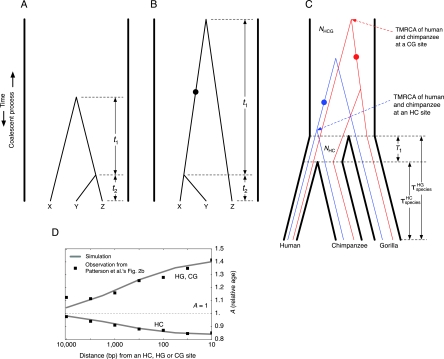
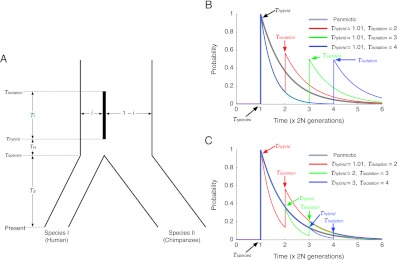
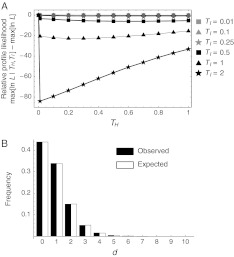
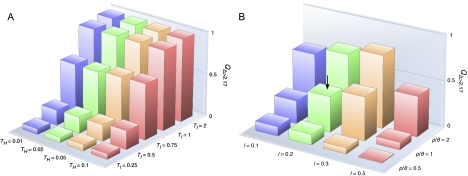
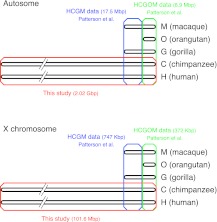
There have been conflicting arguments as to what happened in the human-chimpanzee speciation event. Patterson et al. (2006, Genetic evidence for complex speciation of humans and chimpanzees. Nature 441:1103-1108) proposed a hypothesis that the human-chimpanzee speciation event involved a complicated demographic process: that is, the ancestral lineages of humans and chimpanzees experienced temporal isolation followed by a hybridization event. This hypothesis stemmed from two major observations: a wide range of human-chimpanzee nucleotide divergence across the autosomal genome and very low divergence in the X chromosome. In contrast, Innan and Watanabe (2006, The effect of gene flow on the coalescent time in the human-chimpanzee ancestral population. Mol Biol Evol. 23:1040-1047) demonstrated that the null model of instantaneous speciation fits the genome-wide divergence data for the two species better than alternative models involving partial isolation and migration. To reconcile these two conflicting reports, we first reexamined the analysis of autosomal data by Patterson et al. (2006). By providing a theoretical framework for their analysis, we demonstrated that their observation is what is theoretically expected under the null model of instantaneous speciation with a large ancestral population. Our analysis indicated that the observed wide range of autosomal divergence is simply due to the coalescent process in the large ancestral population of the two species. To further verify this, we developed a maximum likelihood function to detect evidence of hybridization in genome-wide divergence data. Again, the null model with no hybridization best fits the data. We conclude that the simplest speciation model with instantaneous split adequately describes the human-chimpanzee speciation event, and there is no strong reason to involve complicated factors in explaining the autosomal data.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
