

Differential expression in RNA-seq: a matter of depth
- PMID: 21903743
- PMCID: PMC3227109
- DOI: 10.1101/gr.124321.111
Differential expression in RNA-seq: a matter of depth
Abstract
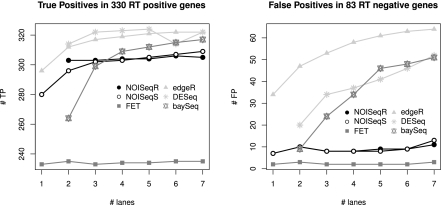
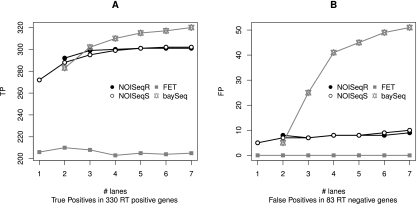
Next-generation sequencing (NGS) technologies are revolutionizing genome research, and in particular, their application to transcriptomics (RNA-seq) is increasingly being used for gene expression profiling as a replacement for microarrays. However, the properties of RNA-seq data have not been yet fully established, and additional research is needed for understanding how these data respond to differential expression analysis. In this work, we set out to gain insights into the characteristics of RNA-seq data analysis by studying an important parameter of this technology: the sequencing depth. We have analyzed how sequencing depth affects the detection of transcripts and their identification as differentially expressed, looking at aspects such as transcript biotype, length, expression level, and fold-change. We have evaluated different algorithms available for the analysis of RNA-seq and proposed a novel approach--NOISeq--that differs from existing methods in that it is data-adaptive and nonparametric. Our results reveal that most existing methodologies suffer from a strong dependency on sequencing depth for their differential expression calls and that this results in a considerable number of false positives that increases as the number of reads grows. In contrast, our proposed method models the noise distribution from the actual data, can therefore better adapt to the size of the data set, and is more effective in controlling the rate of false discoveries. This work discusses the true potential of RNA-seq for studying regulation at low expression ranges, the noise within RNA-seq data, and the issue of replication.
Figures

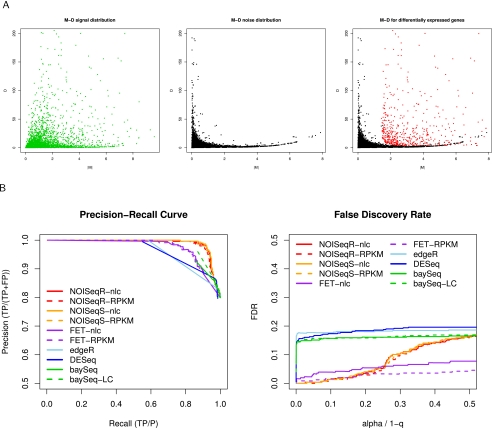
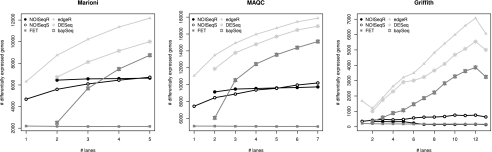
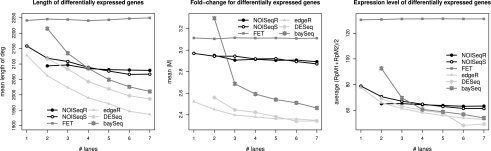
 .
.
References
-
- Anders S 2010. Htseq: analysing high-throughput sequencing data with python. http://www-huber.embl.de/users/anders/HTSeq/ - PMC - PubMed
-
- Anderson J 2005. RNA turnover: unexpected consequences of being tailed. Curr Biol 15: R635–R638 - PubMed
-
- Argout X, Salse J, Aury J, Guiltinan M, Droc G, Gouzy J, Allegre M, Chaparro C, Legavre T, Maximova S, et al. 2010. The genome of Theobroma cacao. Nat Genet 43: 101–108 - PubMed
-
- Blencowe BJ, Ahmad S, Lee LJ 2009. Current-generation high-throughput sequencing: deepening insights into mammalian transcriptomes. Genes Dev 23: 1379–1386 - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources