

GSK3 and Alzheimer's Disease: Facts and Fiction…
- PMID: 21904524
- PMCID: PMC3162188
- DOI: 10.3389/fnmol.2011.00017
GSK3 and Alzheimer's Disease: Facts and Fiction…
Abstract
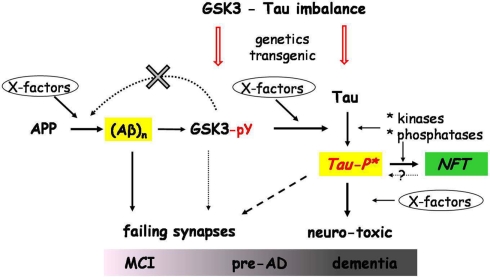
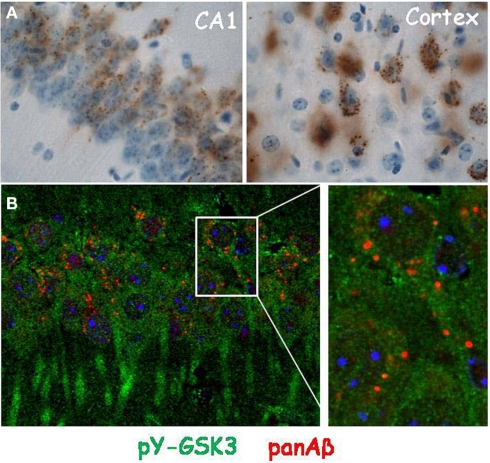
The physiological functions and pathological roles of the Glycogen synthase kinase-type 3 (GSK3) kinases in peripheral and central systems are diverse and complex, and therefore hard to unravel in molecular detail in vivo. Our assignment to review and discuss available data to clarify the actual position of these kinases in the pathology of Alzheimer's dementia (AD) was both ambitious and easy. On the one hand, numerous studies are available in isolated, recombinant, or cell-based systems, which have resulted in very diverse data-sets that are hardly informative for the brain in vivo. At the other extreme, reliable, and relevant models for the role of GSK3 in CNS are rare, if not lacking. Moreover, (too) many in vivo studies used Li(+) as "specific" inhibitor of GSK3, which is factually not valid because lithium ions are neither specific nor potent inhibitors of GSK3 in vivo. More specific pharmacological inhibitors of GSK3 have met with considerable problems, and are reviewed by others in this issue or elsewhere. We concentrate here on AD-related aspects of GSK3 in brain in vivo, mainly studied in transgenic mice and highlight some of the more important issues, among many remaining: activation of GSK3 by amyloid, phosphorylation of protein tau, effects on or interference with synaptic activity, differentiation between both GSK3 isoforms. These relate directly to brain function, and brain dysfunction in AD, and are to be resolved if we want to understand the molecular pathology of this dreadful disease.
Keywords: Alzheimer; GSK3; tau.
Figures
References
-
- Azoulay-Alfaguter I., Yaffe Y., Licht-Murava A., Urbanska M., Jaworski J., Pietrokovski S., Hirschberg K., Eldar-Finkelman H. (2011). Distinct molecular regulation of glycogen synthase kinase-3alpha isozyme controlled by its N-terminal region: functional role in calcium/calpain signaling. J. Biol. Chem. 286, 13470–1348010.1074/jbc.M110.127969 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous