

Use of human stem cell derived cardiomyocytes to examine sunitinib mediated cardiotoxicity and electrophysiological alterations
- PMID: 21906609
- PMCID: PMC5858560
- DOI: 10.1016/j.taap.2011.08.020
Use of human stem cell derived cardiomyocytes to examine sunitinib mediated cardiotoxicity and electrophysiological alterations
Abstract
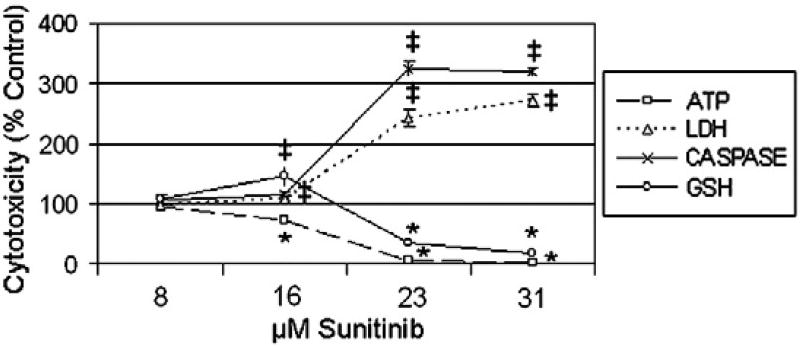
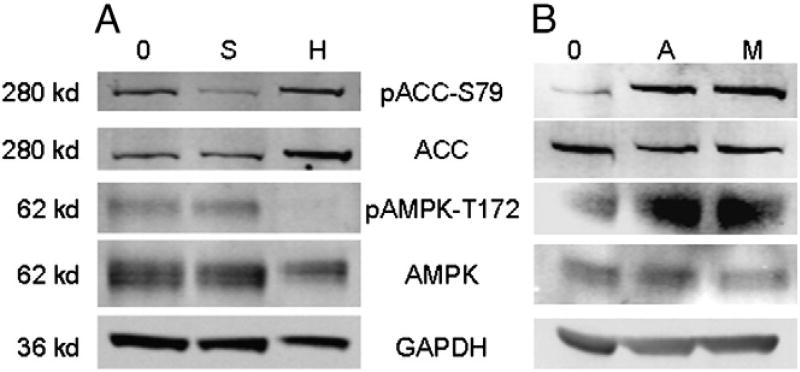
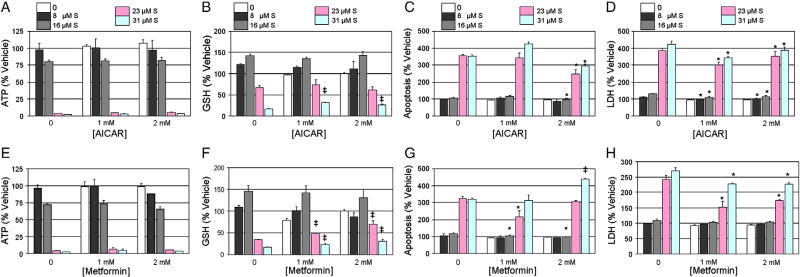
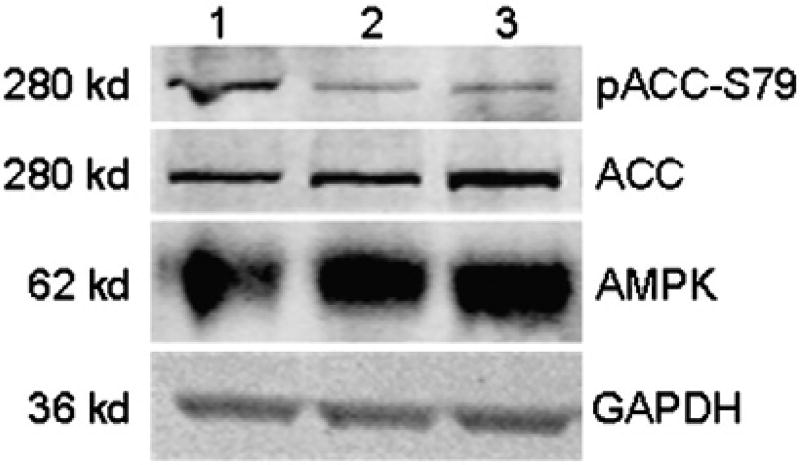
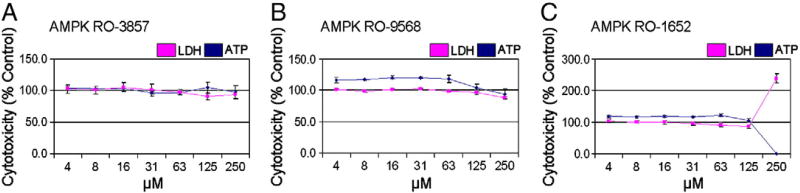
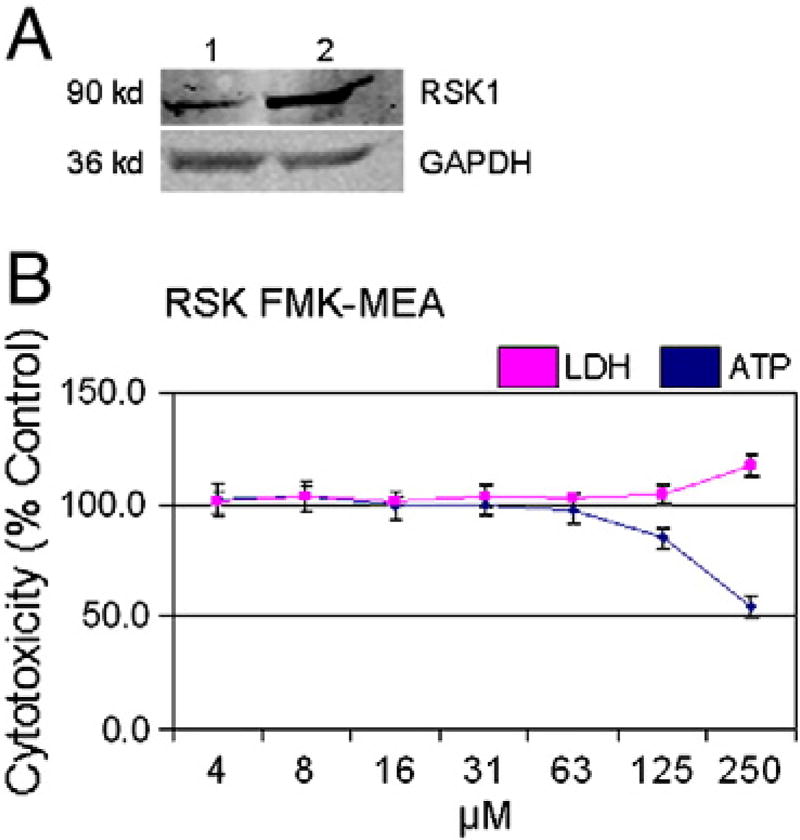
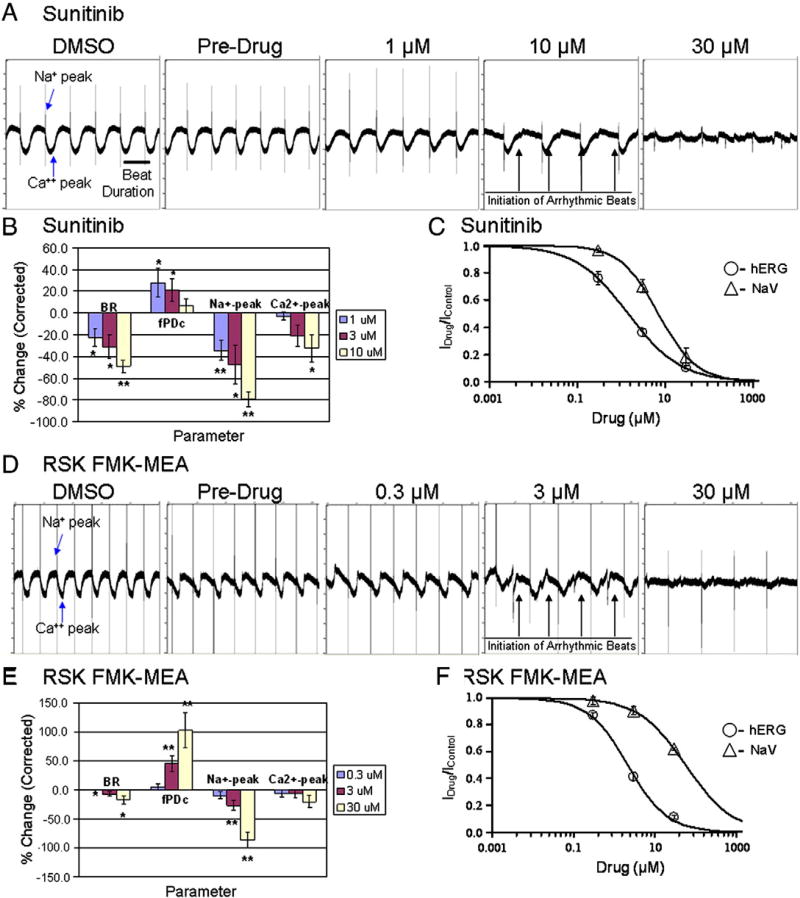
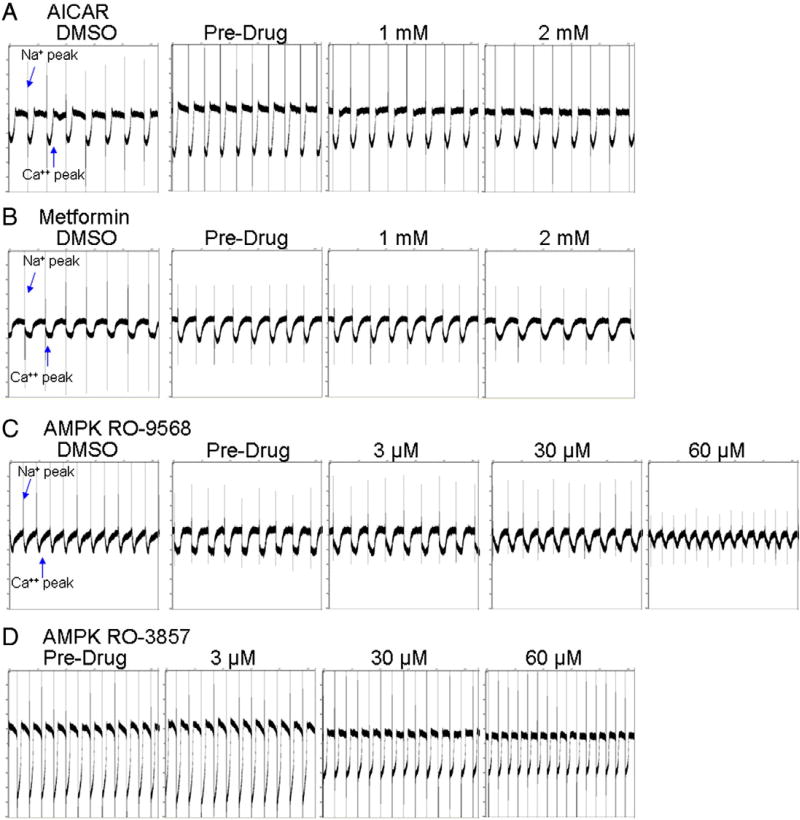
Sunitinib, an oral tyrosine kinase inhibitor approved to treat advanced renal cell carcinoma and gastrointestinal stroma tumor, is associated with clinical cardiac toxicity. Although the precise mechanism of sunitinib cardiotoxicity is not known, both the key metabolic energy regulator, AMP-activated protein kinase (AMPK), and ribosomal S 6 kinase (RSK) have been hypothesized as causative, albeit based on rodent models. To study the mechanism of sunitinib-mediated cardiotoxicity in a human model, induced pluripotent stem cell-derived cardiomyocytes (iPSC-CMs) having electrophysiological and contractile properties of native cardiac tissue were investigated. Sunitinib was cardiotoxic in a dose-dependent manner with an IC₅₀ in the low micromolar range, observed by a loss of cellular ATP, an increase in oxidized glutathione, and induction of apoptosis in iPSC-CMs. Pretreatment of iPSC-CMs with AMPK activators AICAR or metformin, increased the phosphorylation of pAMPK-T172 and pACC-S79, but only marginally attenuated sunitinib mediated cell death. Furthermore, additional inhibitors of AMPK were not directly cytotoxic to iPSC-CMs up to 250 μM concentrations. Inhibition of RSK with a highly specific, irreversible, small molecule inhibitor (RSK-FMK-MEA) did not induce cytotoxicity in iPSC-CMs below 250 μM. Extensive electrophysiological analysis of sunitinib and RSK-FMK-MEA mediated conduction effects were performed. Taken together, these findings suggest that inhibition of AMPK and RSK are not a major component of sunitinib-induced cardiotoxicity. Although the exact mechanism of cardiotoxicity of sunitinib is not known, it is likely due to inhibition of multiple kinases simultaneously. These data highlight the utility of human iPSC-CMs in investigating the potential molecular mechanisms underlying drug-induced cardiotoxicity.
Copyright © 2011 Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Mechanisms of myocyte cytotoxicity induced by the multiple receptor tyrosine kinase inhibitor sunitinib.Mol Pharmacol. 2008 Dec;74(6):1722-8. doi: 10.1124/mol.108.050104. Epub 2008 Sep 24. Mol Pharmacol. 2008. PMID: 18815214
-
Hsp90 inhibitor geldanamycin attenuates the cytotoxicity of sunitinib in cardiomyocytes via inhibition of the autophagy pathway.Toxicol Appl Pharmacol. 2017 Aug 15;329:282-292. doi: 10.1016/j.taap.2017.06.015. Epub 2017 Jun 15. Toxicol Appl Pharmacol. 2017. PMID: 28624441
-
Multi-parameter in vitro toxicity testing of crizotinib, sunitinib, erlotinib, and nilotinib in human cardiomyocytes.Toxicol Appl Pharmacol. 2013 Oct 1;272(1):245-55. doi: 10.1016/j.taap.2013.04.027. Epub 2013 May 21. Toxicol Appl Pharmacol. 2013. PMID: 23707608
-
Moving beyond the comprehensive in vitro proarrhythmia assay: Use of human-induced pluripotent stem cell-derived cardiomyocytes to assess contractile effects associated with drug-induced structural cardiotoxicity.J Appl Toxicol. 2018 Sep;38(9):1166-1176. doi: 10.1002/jat.3611. Epub 2018 Feb 27. J Appl Toxicol. 2018. PMID: 29484688 Review.
-
Drug Development and the Use of Induced Pluripotent Stem Cell-Derived Cardiomyocytes for Disease Modeling and Drug Toxicity Screening.Int J Mol Sci. 2020 Oct 3;21(19):7320. doi: 10.3390/ijms21197320. Int J Mol Sci. 2020. PMID: 33023024 Free PMC article. Review.
Cited by
-
Exaggerated Cardiotoxicity of Sunitinib in Stressed 3-Dimensional Heart Muscles.JACC Basic Transl Sci. 2018 May 30;3(2):277-279. doi: 10.1016/j.jacbts.2018.01.011. eCollection 2018 Apr. JACC Basic Transl Sci. 2018. PMID: 30062213 Free PMC article.
-
Human In Vitro Models for Assessing the Genomic Basis of Chemotherapy-Induced Cardiovascular Toxicity.J Cardiovasc Transl Res. 2020 Jun;13(3):377-389. doi: 10.1007/s12265-020-09962-x. Epub 2020 Feb 20. J Cardiovasc Transl Res. 2020. PMID: 32078739 Free PMC article. Review.
-
Modeling Precision Cardio-Oncology: Using Human-Induced Pluripotent Stem Cells for Risk Stratification and Prevention.Curr Oncol Rep. 2021 May 3;23(7):77. doi: 10.1007/s11912-021-01066-2. Curr Oncol Rep. 2021. PMID: 33937943 Free PMC article. Review.
-
Cytotoxicity of propofol in human induced pluripotent stem cell-derived cardiomyocytes.J Anesth. 2018 Feb;32(1):120-131. doi: 10.1007/s00540-017-2441-0. Epub 2017 Dec 29. J Anesth. 2018. PMID: 29288336 Free PMC article.
-
Modeling the blood-brain barrier using stem cell sources.Fluids Barriers CNS. 2013 Jan 10;10(1):2. doi: 10.1186/2045-8118-10-2. Fluids Barriers CNS. 2013. PMID: 23305164 Free PMC article.
References
-
- Ahmad F, Arad M, Musi N, He H, Wolf C, Branco D, Perez-Atayde AR, Stapleton D, Bali D, Xing Y, Tian R, Goodyear LJ, Berul CI, Ingwall JS, Seidman CE, Seidman JG. Increased alpha2 subunit-associated AMPK activity and PRKAG2 cardiomyopathy. Circulation. 2005;112:3140–3148. - PubMed
-
- Anjum R, Blenis J. The RSK family of kinases: emerging roles in cellular signalling. Nat. Rev. Mol. Cell. Biol. 2008;9:747–758. - PubMed
-
- Arad M, Moskowitz IP, Patel VV, Ahmad F, Perez-Atayde AR, Sawyer DB, Walter M, Li GH, Burgon PG, Maguire CT, Stapleton D, Schmitt JP, Guo XX, Pizard A, Kupershmidt S, Roden DM, Berul CI, Seidman CE, Seidman JG. Transgenic mice overexpressing mutant PRKAG2 define the cause of Wolff–Parkinson–White syndrome in glycogen storage cardiomyopathy. Circulation. 2003;107:2850–2856. - PubMed
-
- Bantscheff M, Eberhard D, Abraham Y, Bastuck S, Boesche M, Hobson S, Mathieson T, Perrin J, Raida M, Rau C, Reader V, Sweetman G, Bauer A, Bouwmeester T, Hopf C, Kruse U, Neubauer G, Ramsden N, Rick J, Kuster B, Drewes G. Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat. Biotechnol. 2007;25:1035–1044. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
