

Pathophysiology of Huntington's disease: time-dependent alterations in synaptic and receptor function
- PMID: 21907762
- PMCID: PMC3221774
- DOI: 10.1016/j.neuroscience.2011.08.052
Pathophysiology of Huntington's disease: time-dependent alterations in synaptic and receptor function
Abstract
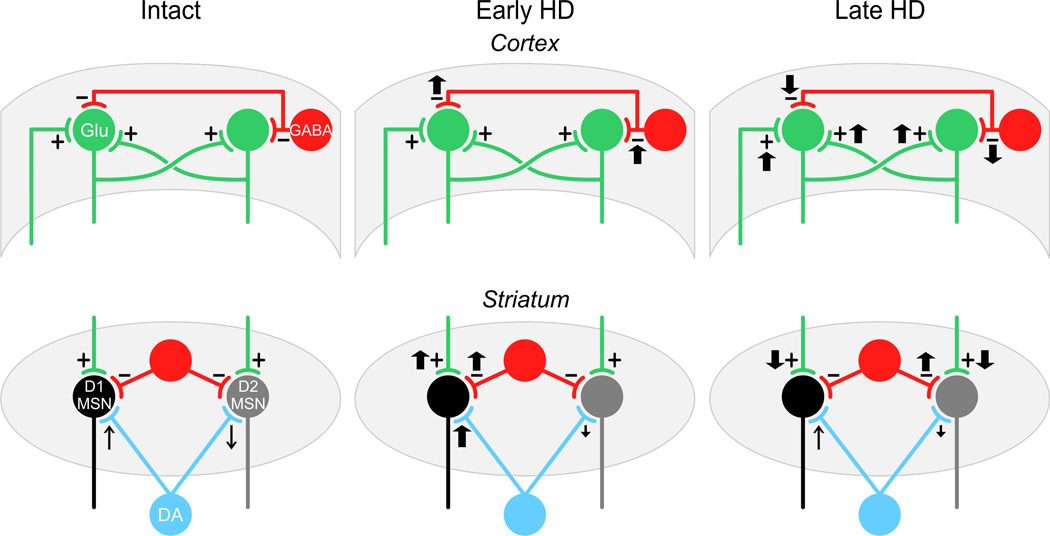
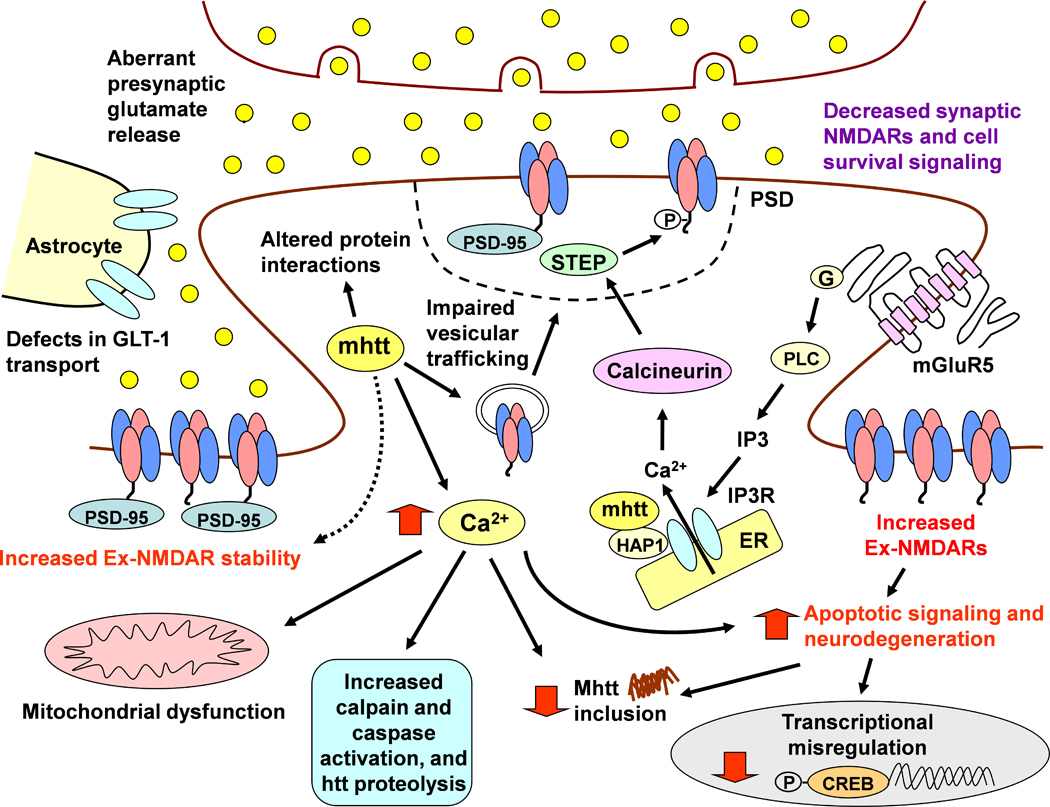
Huntington's disease (HD) is a progressive, fatal neurological condition caused by an expansion of CAG (glutamine) repeats in the coding region of the Huntington gene. To date, there is no cure but great strides have been made to understand pathophysiological mechanisms. In particular, genetic animal models of HD have been instrumental in elucidating the progression of behavioral and physiological alterations, which had not been possible using classic neurotoxin models. Our groups have pioneered the use of transgenic HD mice to examine the excitotoxicity hypothesis of striatal neuronal dysfunction and degeneration, as well as alterations in excitation and inhibition in striatum and cerebral cortex. In this review, we focus on synaptic and receptor alterations of striatal medium-sized spiny (MSNs) and cortical pyramidal neurons in genetic HD mouse models. We demonstrate a complex series of alterations that are region-specific and time-dependent. In particular, many changes are bidirectional depending on the degree of disease progression, that is, early vs. late, and also on the region examined. Early synaptic dysfunction is manifested by dysregulated glutamate release in striatum followed by progressive disconnection between cortex and striatum. The differential effects of altered glutamate release on MSNs originating the direct and indirect pathways is also elucidated, with the unexpected finding that cells of the direct striatal pathway are involved early in the course of the disease. In addition, we review evidence for early N-methyl-D-aspartate receptor (NMDAR) dysfunction leading to enhanced sensitivity of extrasynaptic receptors and a critical role of GluN2B subunits. Some of the alterations in late HD could be compensatory mechanisms designed to cope with early synaptic and receptor dysfunctions. The main findings indicate that HD treatments need to be designed according to the stage of disease progression and should consider regional differences.
Copyright © 2011 IBRO. Published by Elsevier Ltd. All rights reserved.
Figures
References
-
- Albin RL, Reiner A, Anderson KD, Dure LSt, Handelin B, Balfour R, Whetsell WO, Jr, Penney JB, Young AB. Preferential loss of striato-external pallidal projection neurons in presymptomatic Huntington's disease. Ann Neurol. 1992;31:425–430. - PubMed
-
- Albin RL, Reiner A, Anderson KD, Penney JB, Young AB. Striatal and nigral neuron subpopulations in rigid Huntington's disease: implications for the functional anatomy of chorea and rigidity-akinesia. Ann Neurol. 1990a;27:357–365. - PubMed
-
- Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989;12:366–375. - PubMed
-
- Albin RL, Young AB, Penney JB, Handelin B, Balfour R, Anderson KD, Markel DS, Tourtellotte WW, Reiner A. Abnormalities of striatal projection neurons and N-methyl-D-aspartate receptors in presymptomatic Huntington's disease. N Engl J Med. 1990b;322:1293–1298. - PubMed
-
- André VM, Cepeda C, Cummings DM, Jocoy EL, Fisher YE, William Yang X, Levine MS. Dopamine modulation of excitatory currents in the striatum is dictated by the expression of D1 or D2 receptors and modified by endocannabinoids. Eur J Neurosci. 2010a;31:14–28. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
