

Presynaptically silent synapses: dormancy and awakening of presynaptic vesicle release
- PMID: 21908849
- PMCID: PMC3951726
- DOI: 10.1177/1073858411418525
Presynaptically silent synapses: dormancy and awakening of presynaptic vesicle release
Abstract
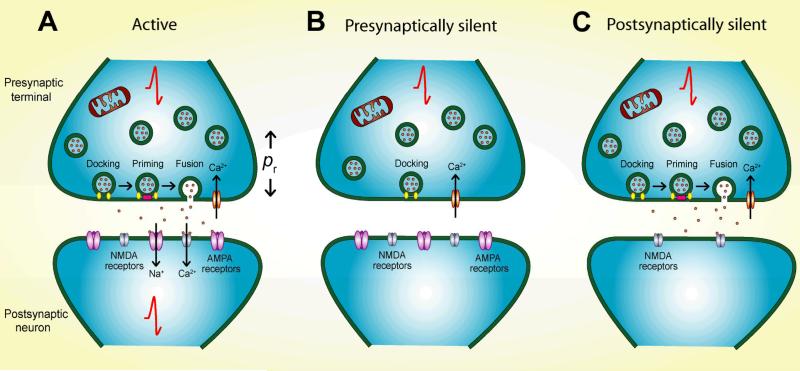
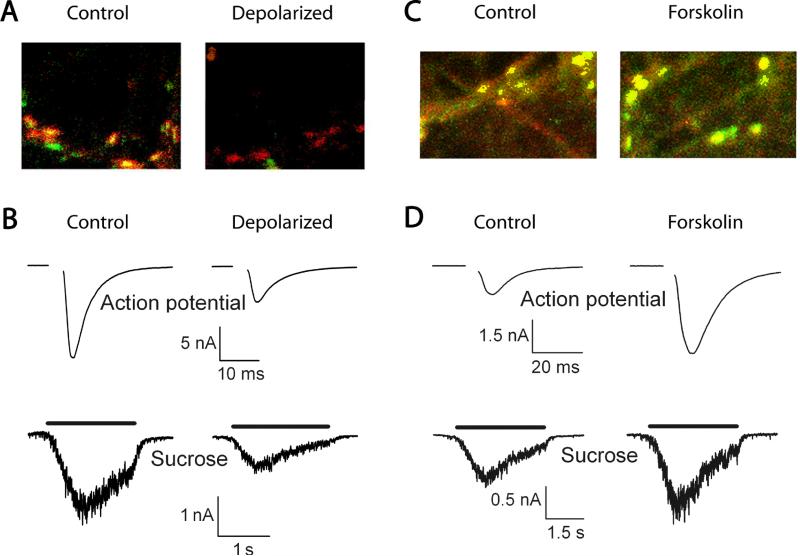
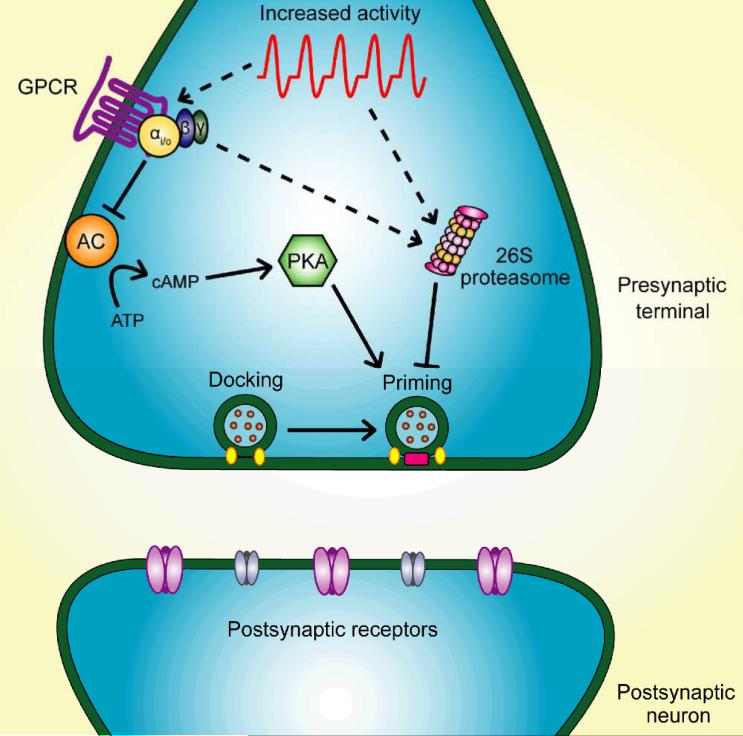
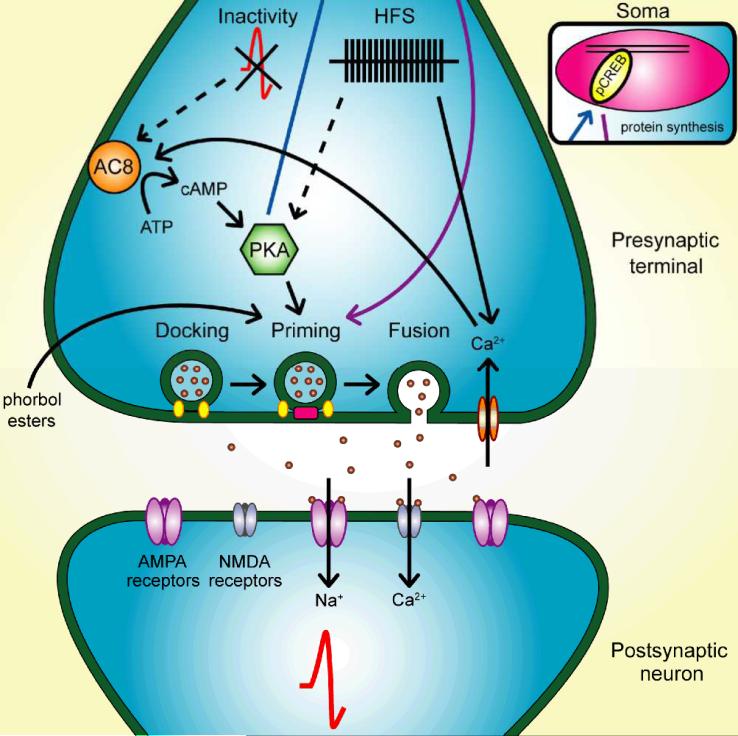
Synapses represent the main junctures of communication between neurons in the nervous system. In many neurotransmitter systems, a fraction of presynaptic terminals fails to release vesicles in response to action potential stimulation and strong calcium influx. These silent presynaptic terminals exhibit a reversible functional dormancy beyond low vesicle release probability, and dormancy status may have important implications in neural function. Recent advances have implicated presynaptic proteins interacting with vesicles downstream of cAMP and protein kinase A signaling cascades in modulating the number of these mute presynaptic terminals, and dormancy induction may represent a homeostatic neuroprotective mechanism active during pathological insults involving excitotoxicity. Interestingly, dormancy reversal may also be induced during Hebbian plasticity. Here, details of synaptic dormancy, recent insights into the molecular signaling cascades involved, and potential clinical and mechanistic implications of this form of synaptic plasticity are described.
Figures
References
-
- Alberini CM, Ghirardi M, Huang YY, Nguyen PV, Kandel ER. A molecular switch for the consolidation of long-term memory: cAMP-inducible gene expression. Ann N Y Acad Sci. 1995;758:261–86. - PubMed
-
- Altrock WD, tom Dieck S, Sokolov M, Meyer AC, Sigler A, Brakebusch C. Functional inactivation of a fraction of excitatory synapses in mice deficient for the active zone protein bassoon. Neuron. 2003;37(5):787–800. others. - PubMed
-
- Augustin I, Rosenmund C, Sudhof TC, Brose N. Munc13-1 is essential for fusion competence of glutamatergic synaptic vesicles. Nature. 1999;400(6743):457–61. - PubMed
-
- Betz A, Ashery U, Rickmann M, Augustin I, Neher E, Sudhof TC. Munc13-1 is a presynaptic phorbol ester receptor that enhances neurotransmitter release. Neuron. 1998;21(1):123–36. others. - PubMed
-
- Bolshakov VY, Golan H, Kandel ER, Siegelbaum SA. Recruitment of new sites of synaptic transmission during the cAMP-dependent late phase of LTP at CA3-CA1 synapses in the hippocampus. Neuron. 1997;19(3):635–51. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
