

Loss of breast epithelial marker hCLCA2 promotes epithelial-to-mesenchymal transition and indicates higher risk of metastasis
- PMID: 21909135
- PMCID: PMC4154589
- DOI: 10.1038/onc.2011.392
Loss of breast epithelial marker hCLCA2 promotes epithelial-to-mesenchymal transition and indicates higher risk of metastasis
Abstract
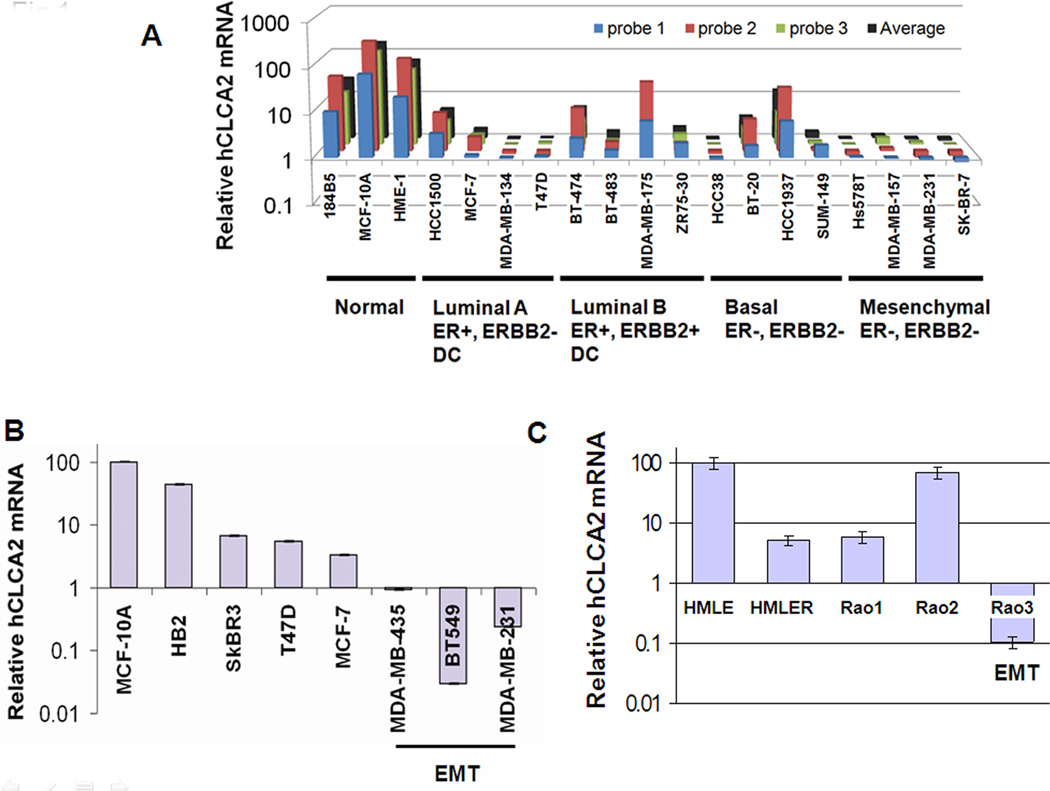
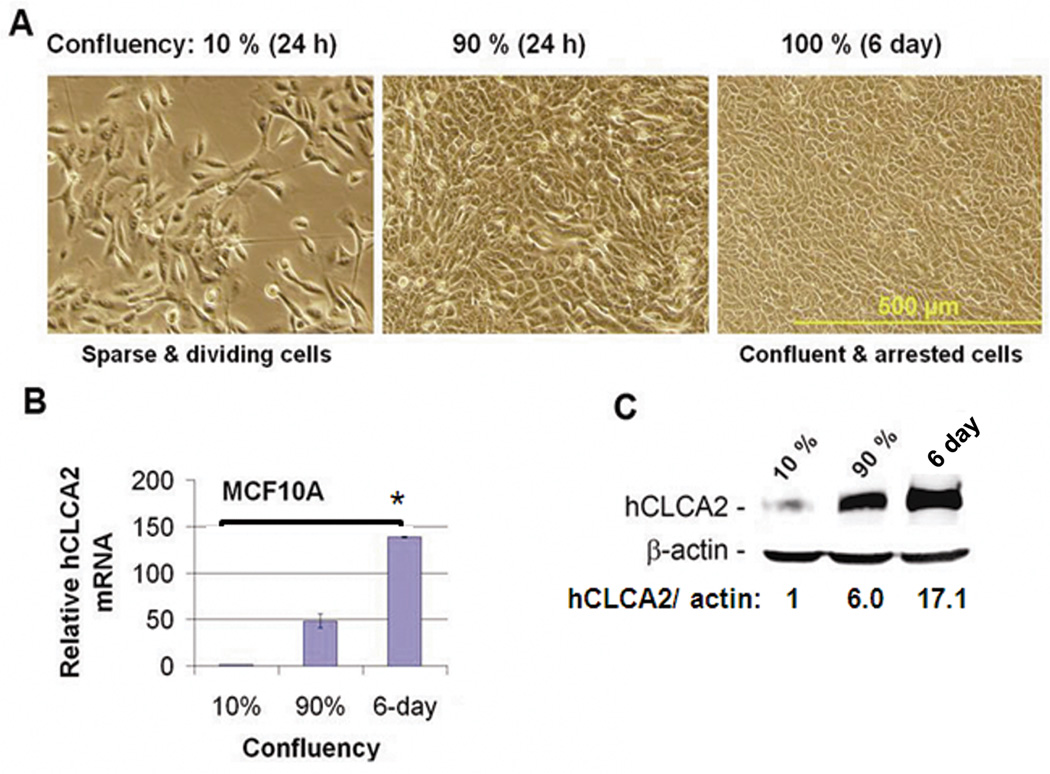
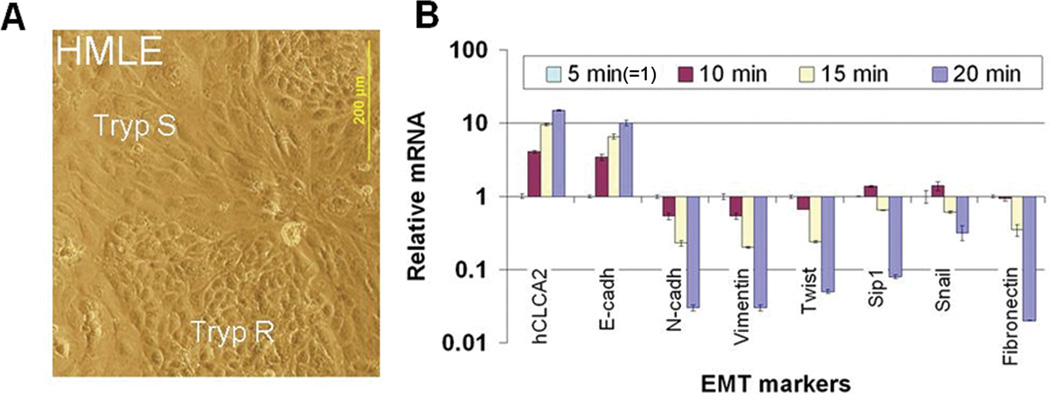
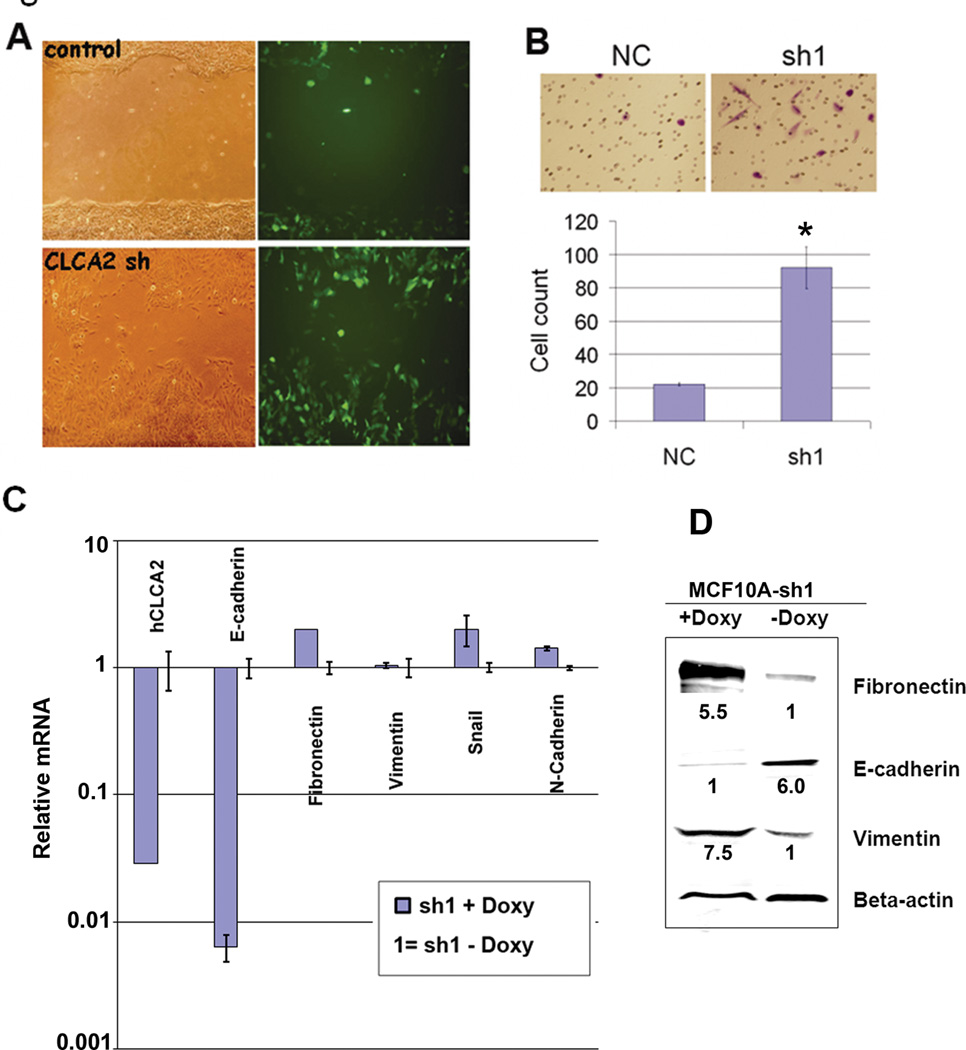
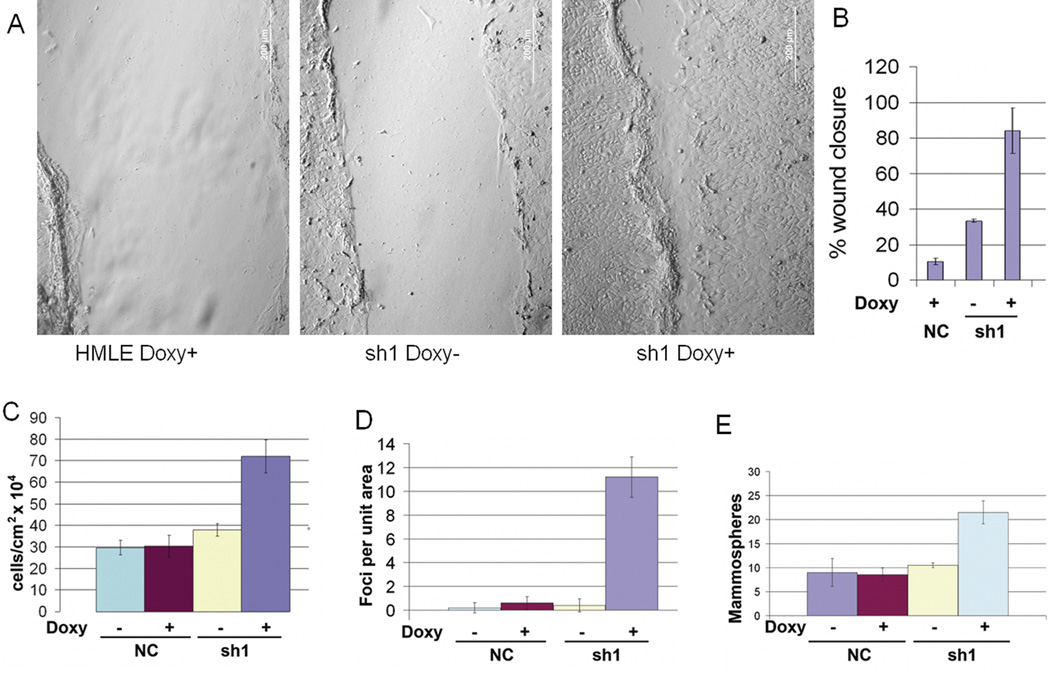
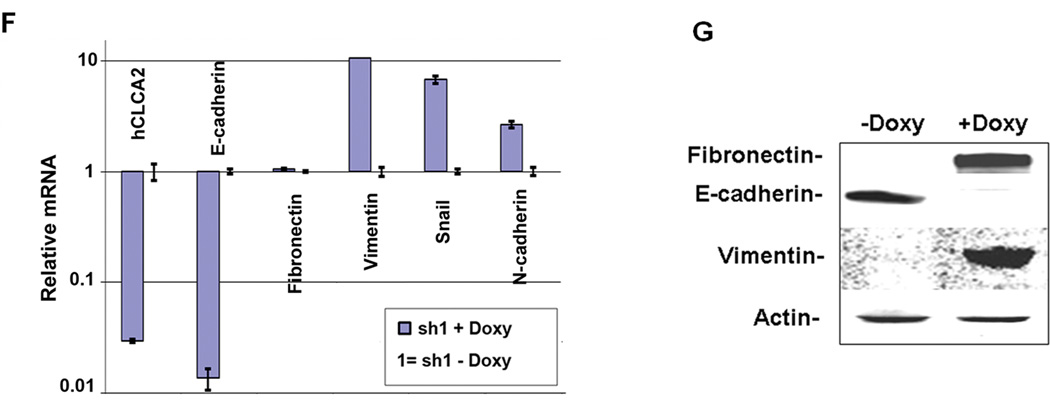
Transition between epithelial and mesenchymal states is a feature of both normal development and tumor progression. We report that expression of chloride channel accessory protein hCLCA2 is a characteristic of epithelial differentiation in the immortalized MCF10A and HMLE models, while induction of epithelial-to-mesenchymal transition by cell dilution, TGFβ or mesenchymal transcription factors sharply reduces hCLCA2 levels. Attenuation of hCLCA2 expression by lentiviral small hairpin RNA caused cell overgrowth and focus formation, enhanced migration and invasion, and increased mammosphere formation in methylcellulose. These changes were accompanied by downregulation of E-cadherin and upregulation of mesenchymal markers such as vimentin and fibronectin. Moreover, hCLCA2 expression is greatly downregulated in breast cancer cells with a mesenchymal or claudin-low profile. These observations suggest that loss of hCLCA2 may promote metastasis. We find that higher-than-median expression of hCLCA2 is associated with a one-third lower rate of metastasis over an 18-year period among breast cancer patients compared with lower-than-median (n=344, unfiltered for subtype). Thus, hCLCA2 is required for epithelial differentiation, and its loss during tumor progression contributes to metastasis. Overexpression of hCLCA2 has been reported to inhibit cell proliferation and is accompanied by increases in chloride current at the plasma membrane and reduced intracellular pH (pHi). We found that knockdown cells have sharply reduced chloride current and higher pHi, both characteristics of tumor cells. These results suggest a mechanism for the effects on differentiation. Loss of hCLCA2 may allow escape from pHi homeostatic mechanisms, permitting the higher intracellular and lower extracellular pH that are characteristic of aggressive tumor cells.
Conflict of interest statement
The authors declare no conflict of interest.
Figures


References
-
- Antonsson B, Conti F, et al. Inhibition of Bax channel-forming activity by Bcl-2. Science. 1997;277(5324):370–372. - PubMed
-
- Barriere H, Poujeol C, et al. CFTR modulates programmed cell death by decreasing intracellular pH in Chinese hamster lung fibroblasts. Am. J. Physiol Cell Physiol. 2001;281(3):C810–C824. - PubMed
-
- Barry MA, Eastman A. Endonuclease activation during apoptosis: the role of cytosolic Ca2+ and pH. Biochem. Biophys. Res. Commun. 1992;186(2):782–789. - PubMed
-
- Beckley JR, Pauli BU, et al. Re-expression of detachment-inducible chloride channel mCLCA5 suppresses growth of metastatic breast cancer cells. J. Biol. Chem. 2004;279(40):41634–41641. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
