

The Trypanosoma cruzi protease cruzain mediates immune evasion
- PMID: 21909255
- PMCID: PMC3164631
- DOI: 10.1371/journal.ppat.1002139
The Trypanosoma cruzi protease cruzain mediates immune evasion
Abstract
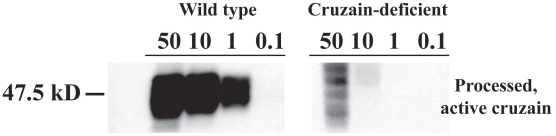
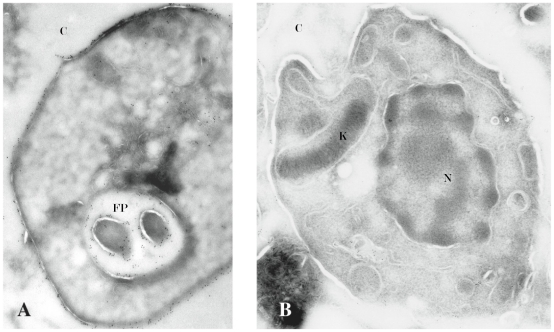
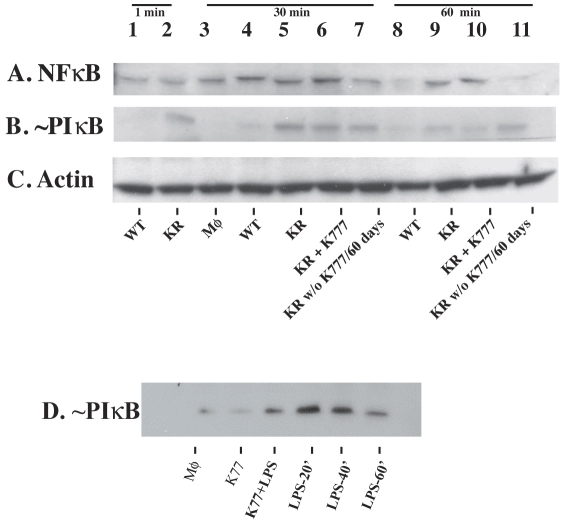
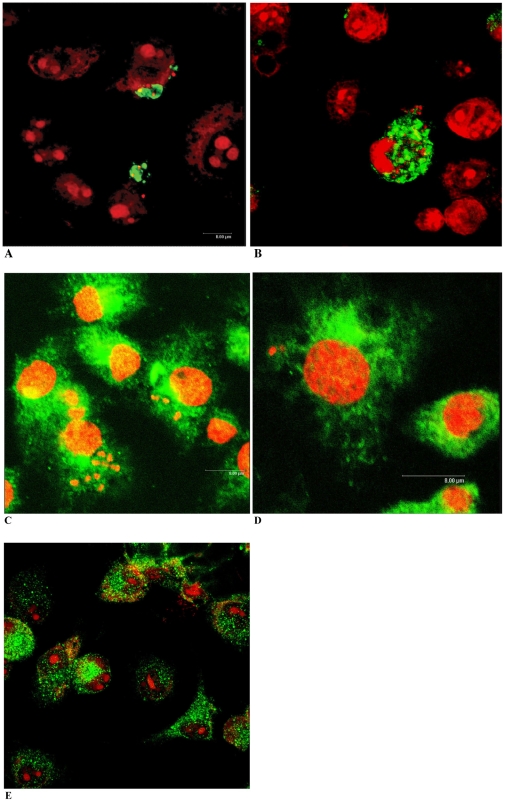
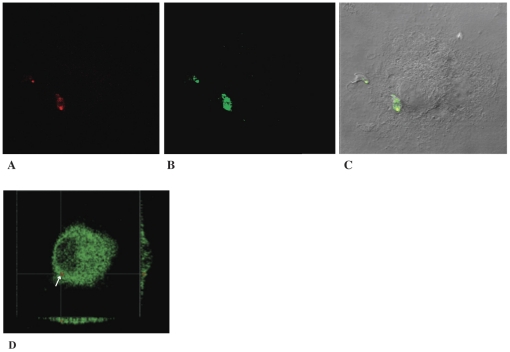
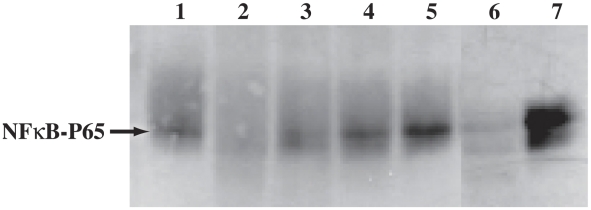
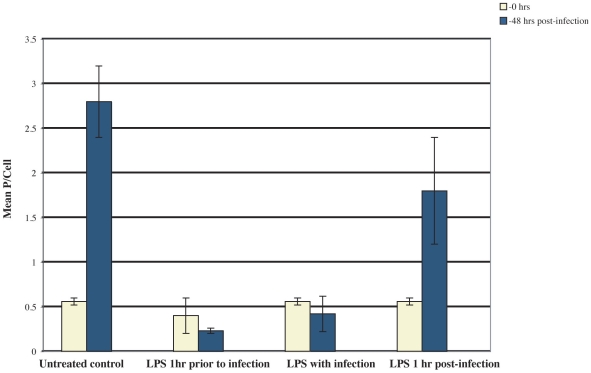
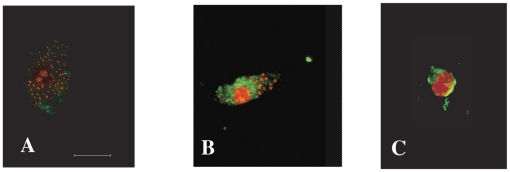
Trypanosoma cruzi is the causative agent of Chagas' disease. Novel chemotherapy with the drug K11777 targets the major cysteine protease cruzain and disrupts amastigote intracellular development. Nevertheless, the biological role of the protease in infection and pathogenesis remains unclear as cruzain gene knockout failed due to genetic redundancy. A role for the T. cruzi cysteine protease cruzain in immune evasion was elucidated in a comparative study of parental wild type- and cruzain-deficient parasites. Wild type T. cruzi did not activate host macrophages during early infection (<60 min) and no increase in ∼P iκB was detected. The signaling factor NF-κB P65 colocalized with cruzain on the cell surface of intracellular wild type parasites, and was proteolytically cleaved. No significant IL-12 expression occurred in macrophages infected with wild type T. cruzi and treated with LPS and BFA, confirming impairment of macrophage activation pathways. In contrast, cruzain-deficient parasites induced macrophage activation, detectable iκB phosphorylation, and nuclear NF-κB P65 localization. These parasites were unable to develop intracellularly and survive within macrophages. IL 12 expression levels in macrophages infected with cruzain-deficient T. cruzi were comparable to LPS activated controls. Thus cruzain hinders macrophage activation during the early (<60 min) stages of infection, by interruption of the NF-κB P65 mediated signaling pathway. These early events allow T. cruzi survival and replication, and may lead to the spread of infection in acute Chagas' disease.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Urbina JA. Specific chemotherapy of Chagas disease: relevance, current limitations and new approaches. Acta Trop. 2010;115:55–68. - PubMed
-
- Chagas C. Coletanea de Trabalhos Cientificos 1909-1913. Editora Universidade de Brasilia. 1982:237–335.
-
- Schuster JP, Schaub GA. Trypanosoma cruzi: skin-penetration kinetics of vector derived metacyclic trypomastigotes. Int J Parasitol. 2000;30:1475–1479. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
