

Impairment of immunoproteasome function by β5i/LMP7 subunit deficiency results in severe enterovirus myocarditis
- PMID: 21909276
- PMCID: PMC3164653
- DOI: 10.1371/journal.ppat.1002233
Impairment of immunoproteasome function by β5i/LMP7 subunit deficiency results in severe enterovirus myocarditis
Abstract
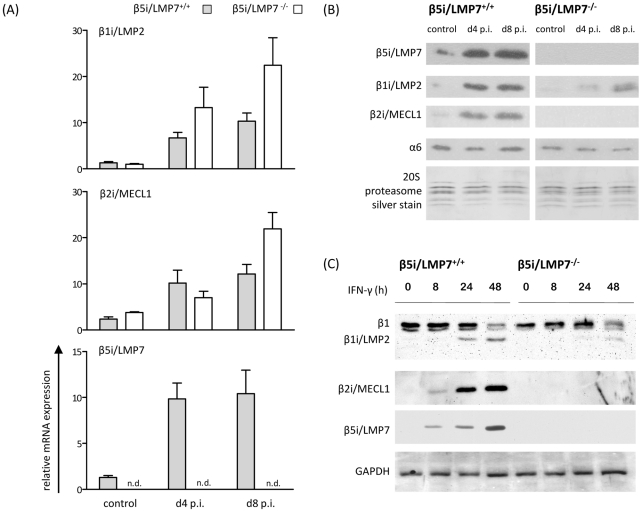
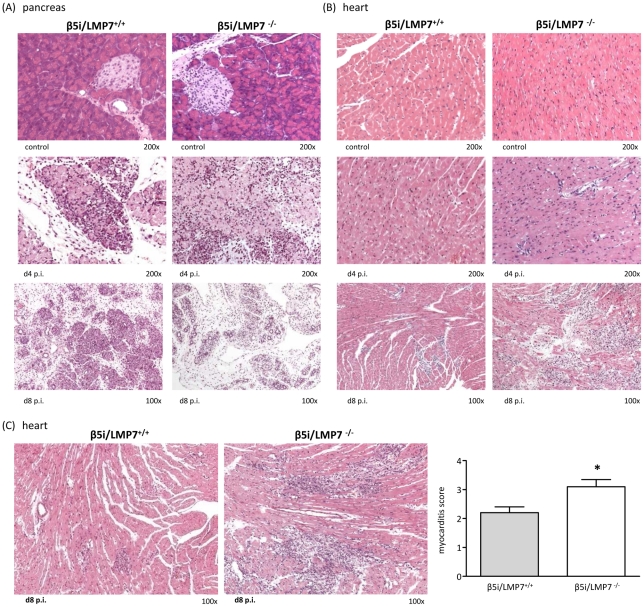
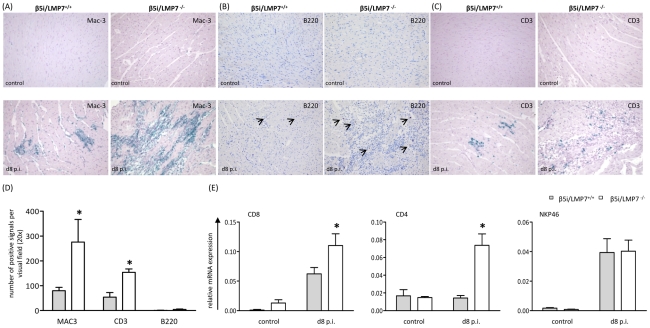
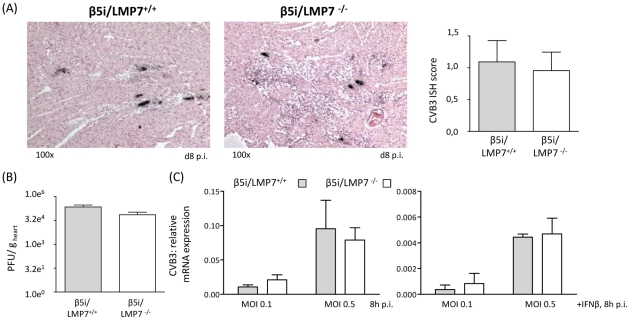
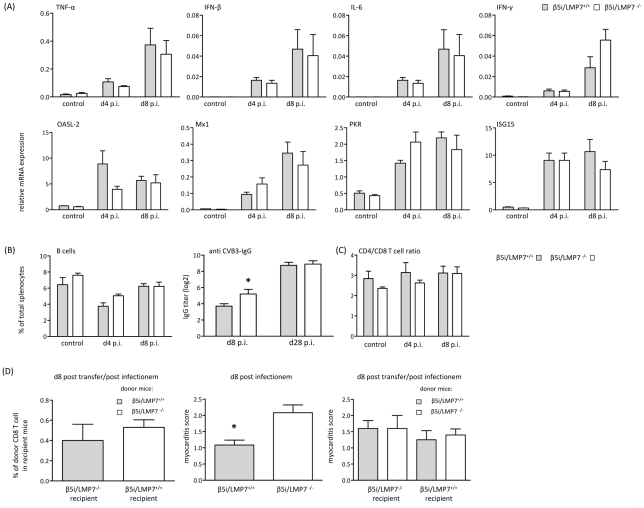
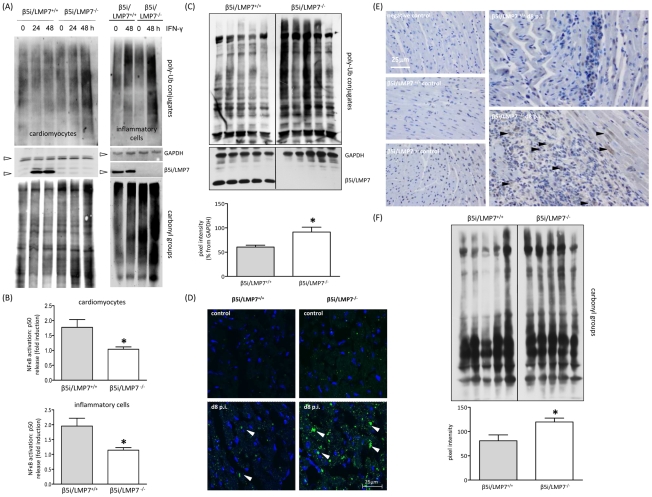
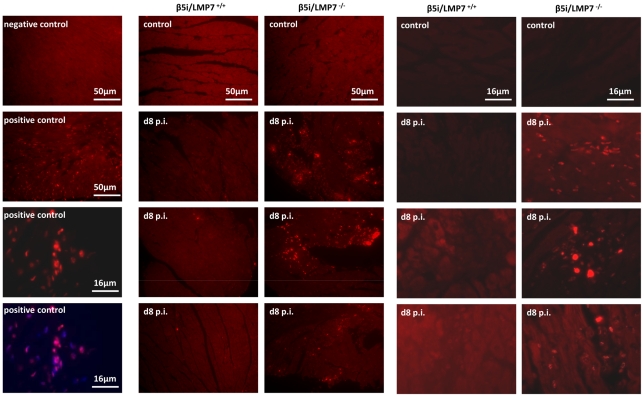
Proteasomes recognize and degrade poly-ubiquitinylated proteins. In infectious disease, cells activated by interferons (IFNs) express three unique catalytic subunits β1i/LMP2, β2i/MECL-1 and β5i/LMP7 forming an alternative proteasome isoform, the immunoproteasome (IP). The in vivo function of IPs in pathogen-induced inflammation is still a matter of controversy. IPs were mainly associated with MHC class I antigen processing. However, recent findings pointed to a more general function of IPs in response to cytokine stress. Here, we report on the role of IPs in acute coxsackievirus B3 (CVB3) myocarditis reflecting one of the most common viral disease entities among young people. Despite identical viral load in both control and IP-deficient mice, IP-deficiency was associated with severe acute heart muscle injury reflected by large foci of inflammatory lesions and severe myocardial tissue damage. Exacerbation of acute heart muscle injury in this host was ascribed to disequilibrium in protein homeostasis in viral heart disease as indicated by the detection of increased proteotoxic stress in cytokine-challenged cardiomyocytes and inflammatory cells from IP-deficient mice. In fact, due to IP-dependent removal of poly-ubiquitinylated protein aggregates in the injured myocardium IPs protected CVB3-challenged mice from oxidant-protein damage. Impaired NFκB activation in IP-deficient cardiomyocytes and inflammatory cells and proteotoxic stress in combination with severe inflammation in CVB3-challenged hearts from IP-deficient mice potentiated apoptotic cell death in this host, thus exacerbating acute tissue damage. Adoptive T cell transfer studies in IP-deficient mice are in agreement with data pointing towards an effective CD8 T cell immune. This study therefore demonstrates that IP formation primarily protects the target organ of CVB3 infection from excessive inflammatory tissue damage in a virus-induced proinflammatory cytokine milieu.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Differential interferon responses enhance viral epitope generation by myocardial immunoproteasomes in murine enterovirus myocarditis.Am J Pathol. 2009 Aug;175(2):510-8. doi: 10.2353/ajpath.2009.090033. Epub 2009 Jul 9. Am J Pathol. 2009. PMID: 19590042 Free PMC article.
-
PA28 modulates antigen processing and viral replication during coxsackievirus B3 infection.PLoS One. 2017 Mar 9;12(3):e0173259. doi: 10.1371/journal.pone.0173259. eCollection 2017. PLoS One. 2017. PMID: 28278207 Free PMC article.
-
Ongoing coxsackievirus myocarditis is associated with increased formation and activity of myocardial immunoproteasomes.Am J Pathol. 2006 May;168(5):1542-52. doi: 10.2353/ajpath.2006.050865. Am J Pathol. 2006. PMID: 16651621 Free PMC article.
-
Emerging roles of immunoproteasomes beyond MHC class I antigen processing.Cell Mol Life Sci. 2012 Aug;69(15):2543-58. doi: 10.1007/s00018-012-0938-0. Epub 2012 Mar 2. Cell Mol Life Sci. 2012. PMID: 22382925 Free PMC article. Review.
-
On the Role of the Immunoproteasome in Protein Homeostasis.Cells. 2021 Nov 18;10(11):3216. doi: 10.3390/cells10113216. Cells. 2021. PMID: 34831438 Free PMC article. Review.
Cited by
-
Regulation of immunoproteasome function in the lung.Sci Rep. 2015 May 19;5:10230. doi: 10.1038/srep10230. Sci Rep. 2015. PMID: 25989070 Free PMC article.
-
Immunoproteasome Inhibition Ameliorates Aged Dystrophic Mouse Muscle Environment.Int J Mol Sci. 2022 Nov 24;23(23):14657. doi: 10.3390/ijms232314657. Int J Mol Sci. 2022. PMID: 36498987 Free PMC article.
-
Aging attenuates redox adaptive homeostasis and proteostasis in female mice exposed to traffic-derived nanoparticles ('vehicular smog').Free Radic Biol Med. 2018 Jun;121:86-97. doi: 10.1016/j.freeradbiomed.2018.04.574. Epub 2018 Apr 27. Free Radic Biol Med. 2018. PMID: 29709705 Free PMC article.
-
Mitigated viral myocarditis in A/J mice by the immunoproteasome inhibitor ONX 0914 depends on inhibition of systemic inflammatory responses in CoxsackievirusB3 infection.Basic Res Cardiol. 2021 Feb 1;116(1):7. doi: 10.1007/s00395-021-00848-w. Basic Res Cardiol. 2021. PMID: 33523326 Free PMC article.
-
Role of Proteasomes in Inflammation.J Clin Med. 2021 Apr 20;10(8):1783. doi: 10.3390/jcm10081783. J Clin Med. 2021. PMID: 33923887 Free PMC article. Review.
References
-
- Watanabe Y, Suzuki O, Haruyama T, Akaike T. Interferon-gamma induces reactive oxygen species and endoplasmic reticulum stress at the hepatic apoptosis. J Cell Biochem. 2003;89:244–253. - PubMed
-
- Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003;426:895–899. - PubMed
-
- Herrmann J, Soares SM, Lerman LO, Lerman A. Potential role of the ubiquitin-proteasome system in atherosclerosis. J Am Coll Cardiol. 2008;51:2003–2010. - PubMed
-
- Patterson C, Ike C, Willis PW, Stouffer GA, Willis MS. The bitter end: the ubiquitin-proteasome system and cardiac dysfunction. Circulation. 2007;115:1456–1463. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
