

The K+ channel opener 1-EBIO potentiates residual function of mutant CFTR in rectal biopsies from cystic fibrosis patients
- PMID: 21909392
- PMCID: PMC3164200
- DOI: 10.1371/journal.pone.0024445
The K+ channel opener 1-EBIO potentiates residual function of mutant CFTR in rectal biopsies from cystic fibrosis patients
Abstract
Background: The identification of strategies to improve mutant CFTR function remains a key priority in the development of new treatments for cystic fibrosis (CF). Previous studies demonstrated that the K⁺ channel opener 1-ethyl-2-benzimidazolone (1-EBIO) potentiates CFTR-mediated Cl⁻ secretion in cultured cells and mouse colon. However, the effects of 1-EBIO on wild-type and mutant CFTR function in native human colonic tissues remain unknown.
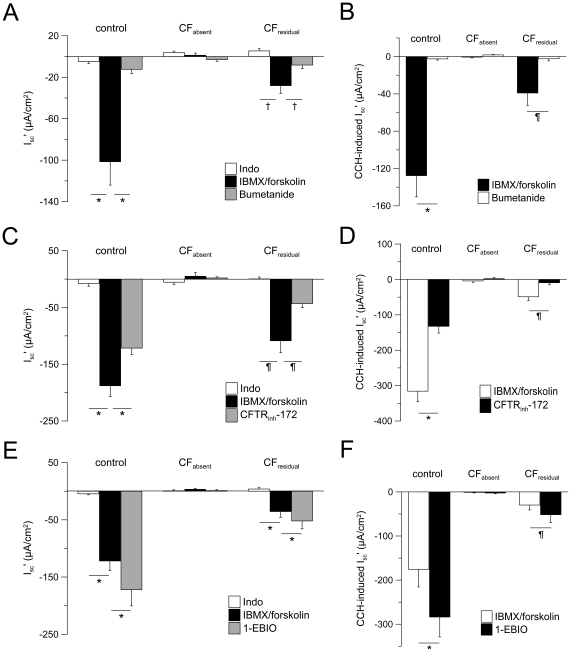
Methods: We studied the effects of 1-EBIO on CFTR-mediated Cl⁻ secretion in rectal biopsies from 47 CF patients carrying a wide spectrum of CFTR mutations and 57 age-matched controls. Rectal tissues were mounted in perfused micro-Ussing chambers and the effects of 1-EBIO were compared in control tissues, CF tissues expressing residual CFTR function and CF tissues with no detectable Cl⁻ secretion.
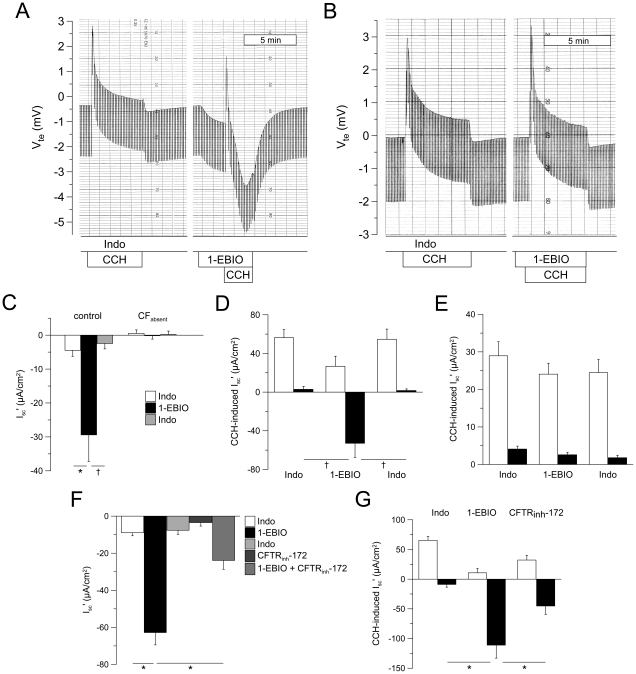
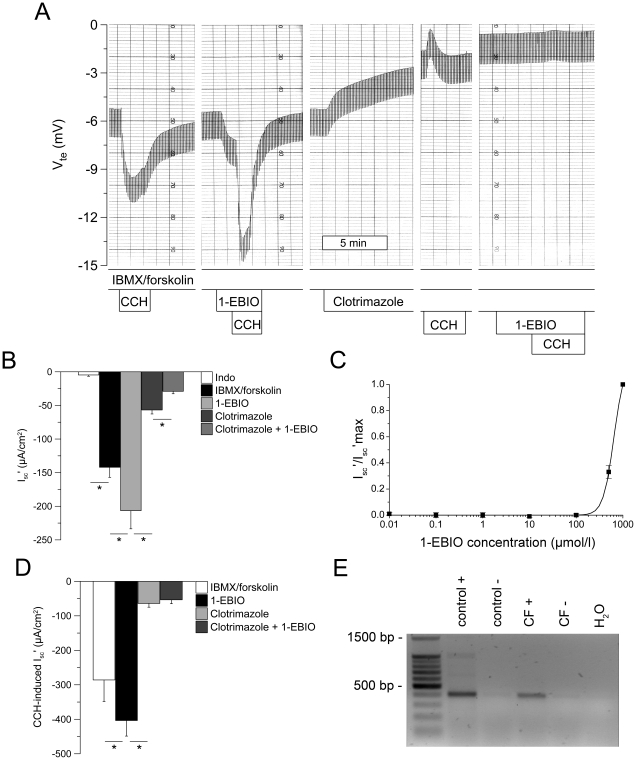
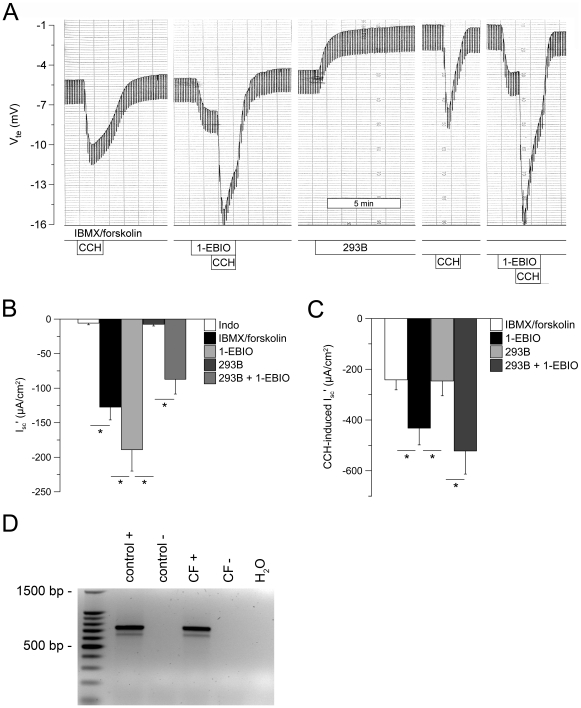
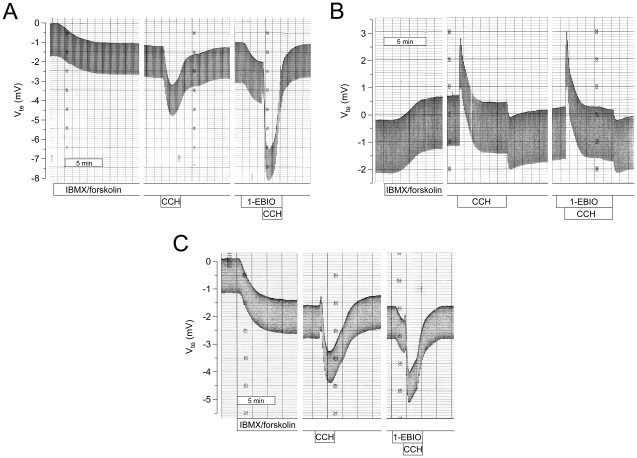
Results: Studies in control tissues demonstrate that 1-EBIO activated CFTR-mediated Cl⁻ secretion in the absence of cAMP-mediated stimulation and potentiated cAMP-induced Cl⁻ secretion by 39.2±6.7% (P<0.001) via activation of basolateral Ca²⁺-activated and clotrimazole-sensitive KCNN4 K⁺ channels. In CF specimens, 1-EBIO potentiated cAMP-induced Cl⁻ secretion in tissues with residual CFTR function by 44.4±11.5% (P<0.001), but had no effect on tissues lacking CFTR-mediated Cl⁻ conductance.
Conclusions: We conclude that 1-EBIO potentiates Cl⁻secretion in native CF tissues expressing CFTR mutants with residual Cl⁻ channel function by activation of basolateral KCNN4 K⁺ channels that increase the driving force for luminal Cl⁻ exit. This mechanism may augment effects of CFTR correctors and potentiators that increase the number and/or activity of mutant CFTR channels at the cell surface and suggests KCNN4 as a therapeutic target for CF.
Conflict of interest statement
Figures
References
-
- Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, et al. Identification of the cystic fibrosis gene: genetic analysis. Science. 1989;245:1073–1080. - PubMed
-
- Welsh MJ, Ramsey BW, Accurso F, Cutting GR. Cystic fibrosis. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic & Molecular Bases of Inherited Disease. 8th ed. New York: McGraw-Hill; 2001. pp. 5121–5188.
-
- Riordan JR. CFTR function and prospects for therapy. Annu Rev Biochem. 2008;77:701–726. - PubMed
-
- Cheng SH, Gregory RJ, Marshall J, Paul S, Souza DW, et al. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell. 1990;63:827–834. - PubMed
-
- Hamosh A, Trapnell BC, Zeitlin PL, Montrose-Rafizadeh C, Rosenstein BJ, et al. Severe deficiency of cystic fibrosis transmembrane conductance regulator messenger RNA carrying nonsense mutations R553X and W1316X in respiratory epithelial cells of patients with cystic fibrosis. J Clin Invest. 1991;88:1880–1885. - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
