

The link between the metabolic syndrome and cancer
- PMID: 21912508
- PMCID: PMC3164150
- DOI: 10.7150/ijbs.7.1003
The link between the metabolic syndrome and cancer
Abstract
Since the incidence of the metabolic syndrome is on the rise in the western world, its coherence to cancer is becoming more apparent. In this review we discuss the different potential factors involved in the increase of cancer in the metabolic syndrome including obesity, dyslipidemia and Type 2 Diabetes Mellitus (T2DM) as well as inflammation and hypoxia. We especially focus on the insulin and IGF systems with their intracellular signaling cascades mediated by different receptor subtypes, and suggest that they may play major roles in this process. Understanding the mechanisms involved will be helpful in developing potential therapeutics.
Keywords: cancer; metabolic syndrome.
Conflict of interest statement
Conflict of Interests: The authors have declared that no conflict of interest exists.
Figures
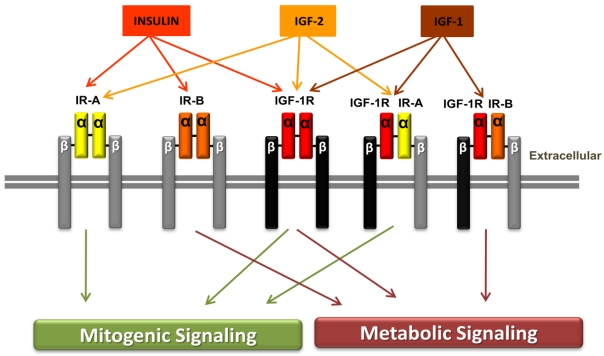
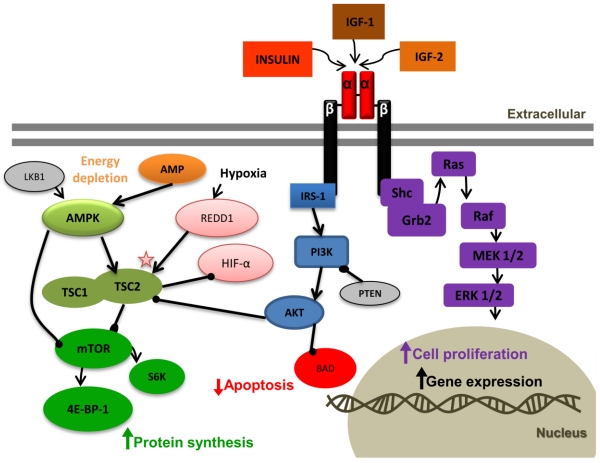
 The detailed interaction between REDD1 and TSC is not clear yet. We conclude that the inhibition of HIF-α by TSC might be independent of REDD1. The mitogen-activated protein kinase (MAPK) pathway can also be activated by IGF-1R activation. In this pathway IGF-1R activates the adaptor proteins, Shc and Grb2, leading to activation of Ras, Raf, MEK1/2, and ERK1/2, which results in cell proliferation.
The detailed interaction between REDD1 and TSC is not clear yet. We conclude that the inhibition of HIF-α by TSC might be independent of REDD1. The mitogen-activated protein kinase (MAPK) pathway can also be activated by IGF-1R activation. In this pathway IGF-1R activates the adaptor proteins, Shc and Grb2, leading to activation of Ras, Raf, MEK1/2, and ERK1/2, which results in cell proliferation.References
-
- Zimmet PZ, Alberti KG. Introduction: Globalization and the non-communicable disease epidemic. Obesity (Silver Spring) 2006;14:1–3. - PubMed
-
- Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med. 2003;348:1625–38. - PubMed
-
- Gallagher EJ NR, Yakar S. The Increased Risk of Cancer in Obesity and Type 2 Diabetes: Potential Mechanisms; Principles of Diabetes Mellitus, 2nd ed. New York, USA: Springer; 2010. pp. 579–99.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
