

Integrated genome-wide analysis of transcription factor occupancy, RNA polymerase II binding and steady-state RNA levels identify differentially regulated functional gene classes
- PMID: 21914722
- PMCID: PMC3245935
- DOI: 10.1093/nar/gkr720
Integrated genome-wide analysis of transcription factor occupancy, RNA polymerase II binding and steady-state RNA levels identify differentially regulated functional gene classes
Abstract
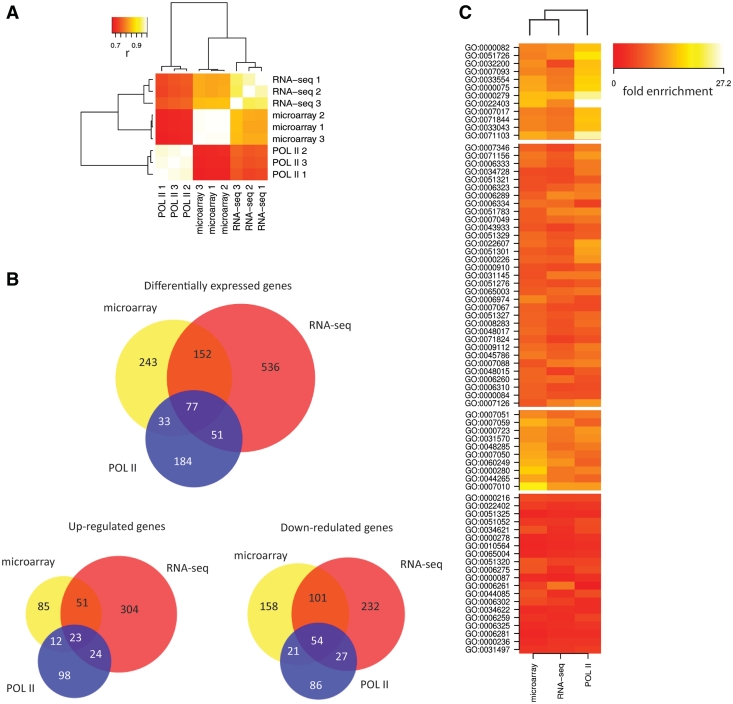
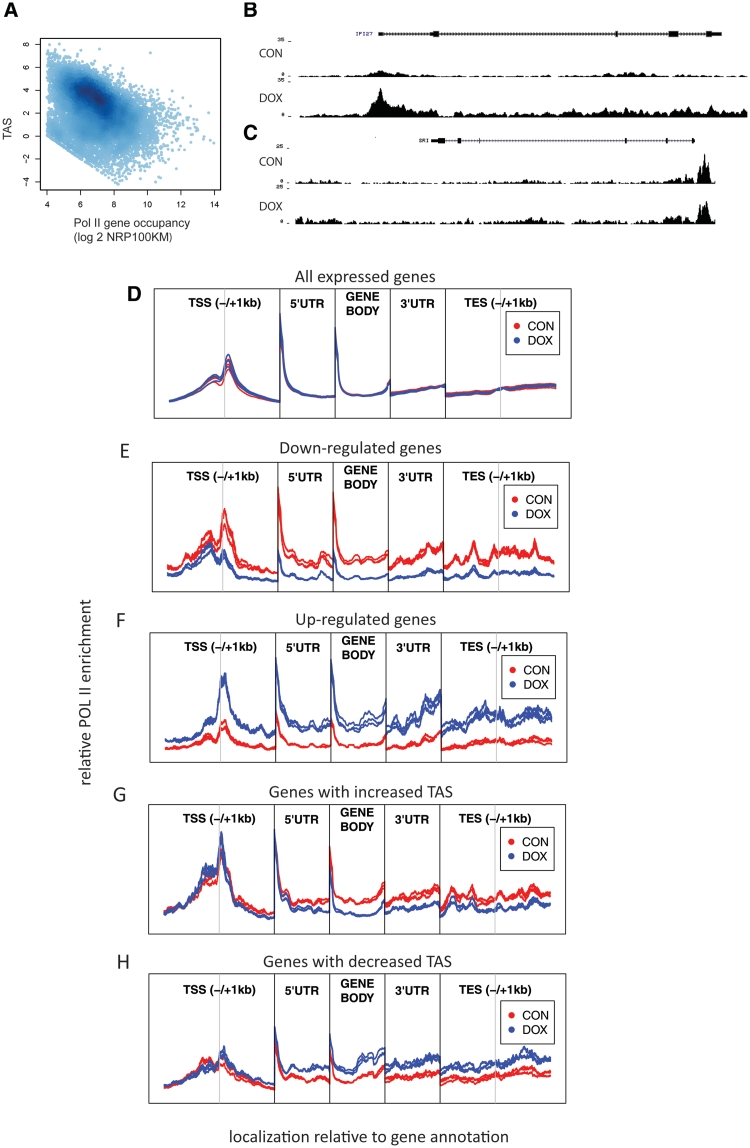
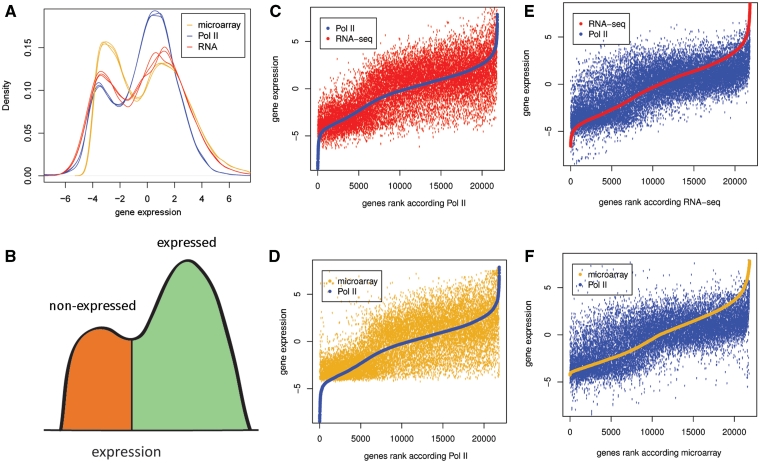
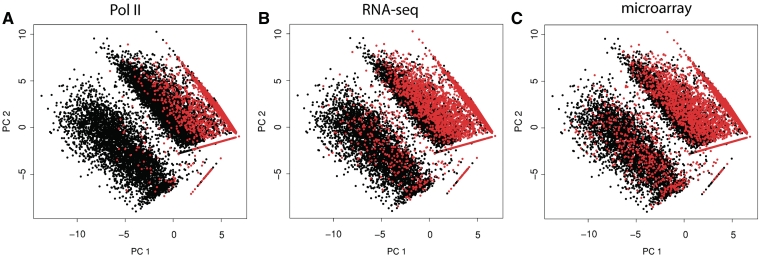
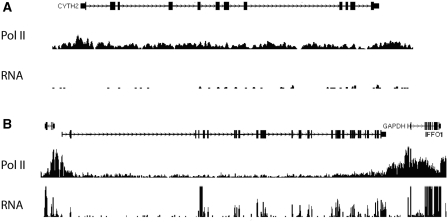
Routine methods for assaying steady-state mRNA levels such as RNA-seq and micro-arrays are commonly used as readouts to study the role of transcription factors (TFs) in gene expression regulation. However, cellular RNA levels do not solely depend on activity of TFs and subsequent transcription by RNA polymerase II (Pol II), but are also affected by RNA turnover rate. Here, we demonstrate that integrated analysis of genome-wide TF occupancy, Pol II binding and steady-state RNA levels provide important insights in gene regulatory mechanisms. Pol II occupancy, as detected by Pol II ChIP-seq, was found to correlate better with TF occupancy compared to steady-state RNA levels and is thus a more precise readout for the primary transcriptional mechanisms that are triggered by signal transduction. Furthermore, analysis of differential Pol II occupancy and RNA-seq levels identified genes with high Pol II occupancy and relatively low RNA levels and vice versa. These categories are strongly enriched for genes from different functional classes. Our results demonstrate a complementary value in Pol II chip-seq and RNA-seq approaches for better understanding of gene expression regulation.
Figures

References
-
- Garcia-Martinez J, Aranda A, Perez-Ortin JE. Genomic run-on evaluates transcription rates for all yeast genes and identifies gene regulatory mechanisms. Mol. Cell. 2004;15:303–313. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
