

Genome-wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa
- PMID: 21915109
- PMCID: PMC3195253
- DOI: 10.1038/ncomms1467
Genome-wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa
Abstract
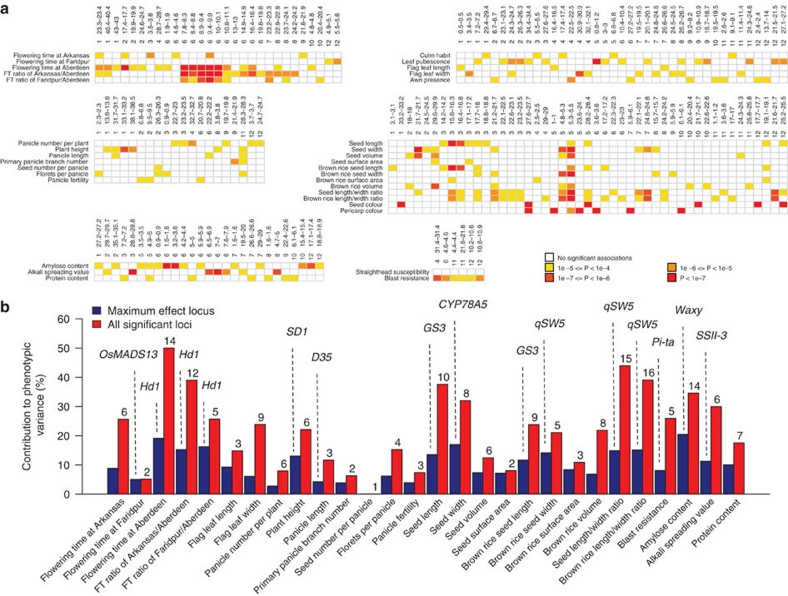
Asian rice, Oryza sativa is a cultivated, inbreeding species that feeds over half of the world's population. Understanding the genetic basis of diverse physiological, developmental, and morphological traits provides the basis for improving yield, quality and sustainability of rice. Here we show the results of a genome-wide association study based on genotyping 44,100 SNP variants across 413 diverse accessions of O. sativa collected from 82 countries that were systematically phenotyped for 34 traits. Using cross-population-based mapping strategies, we identified dozens of common variants influencing numerous complex traits. Significant heterogeneity was observed in the genetic architecture associated with subpopulation structure and response to environment. This work establishes an open-source translational research platform for genome-wide association studies in rice that directly links molecular variation in genes and metabolic pathways with the germplasm resources needed to accelerate varietal development and crop improvement.
Figures





References
-
- Toriyama K., Heong K. L. & Hardy B. Rice is Life: Scientific Perspectives for the 21st Century: Proceedings of the World Rice Research Conference, Tsukuba, Japan (International rice research institute, 2005).
-
- Greenland D. J. The Sustainability of Rice Farming (Cab International, 1997).
-
- Goff S. A. et al.. A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science 296, 92–100 (2002). - PubMed
-
- Yu J. et al.. A draft sequence of the rice genome (Oryza sativa L. ssp. indica). Science 296, 79–92 (2002). - PubMed
-
- Huang X. et al.. Genome-wide association studies of 14 agronomic traits in rice landraces. Nat. Genet. 42, 961–967 (2010). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
