

doi: 10.1186/gb-2011-12-9-127.
The promise and limitations of population exomics for human evolution studies
Affiliations
- PMID: 21920050
- PMCID: PMC3308040
- DOI: 10.1186/gb-2011-12-9-127
Item in Clipboard
The promise and limitations of population exomics for human evolution studies
Genome Biol.
.
Abstract
Exome sequencing is poised to yield substantial insights into human genetic variation and evolutionary history, but there are significant challenges to overcome before this becomes a reality.
Figures
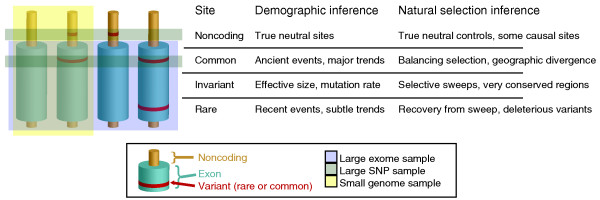
The contributions of different data types to population genetic inferences. Exomes, SNP arrays, and genomes are likely to capture different combinations of these four basic site types: common variants, rare variants, invariant sites, and noncoding sites. Each site type offers unique information relevant to analyses of demography and natural selection. Rare variants and invariant sites captured in exomes are important for numerous evolutionary questions, from estimating the effective population size to detecting positive selective sweeps, and they may be missed by other methods. However, phenotypically causal noncoding variants and truly neutral regions far from genes and free from the effects of selection are absent from exomes.
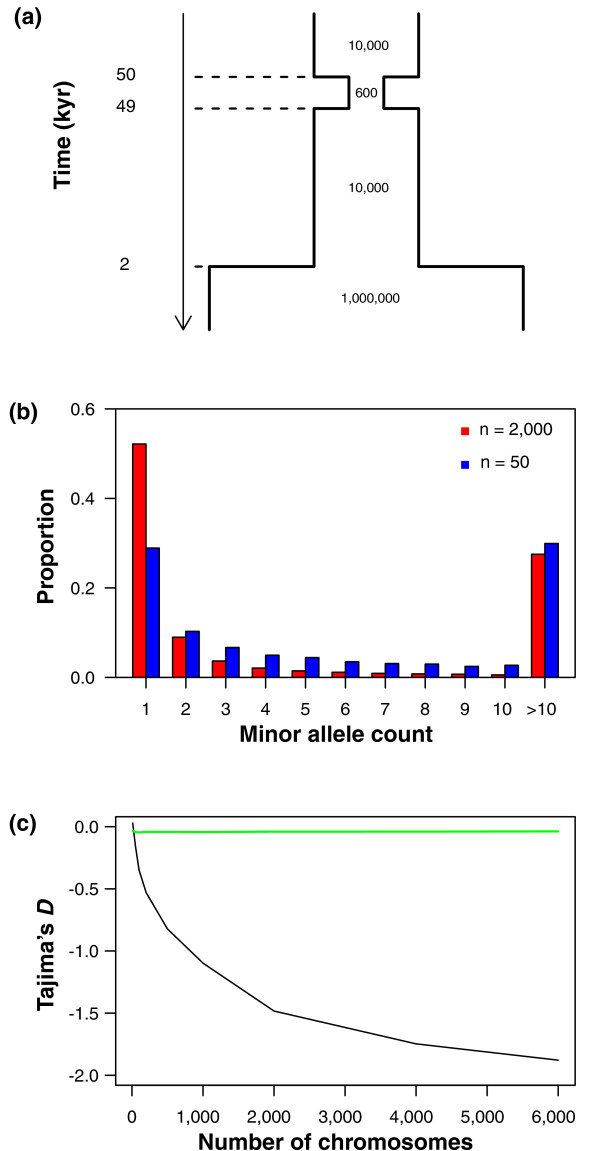
The influence of sample size in detecting recent population expansions. (a) Demographic model used in the simulations. Times of demographic events (in units of thousands of years, kyr) and population sizes are indicated. (b) The site frequency spectrum for sample sizes of 50 and 2,000 chromosomes. (c) Average value of Tajima's D based on 104 simulation replicates as a function of sample size (number of chromosomes). The lines denote values of Tajima's D from the demographic model in (a) (black line) and a constant sized population (green line). This simulation illustrates the power of large sample sizes for inferring recent demographic events. All information in (b, c) is based on simulating 10 kb of sequence.
Similar articles
-
Evaluation of whole exome sequencing as an alternative to BeadChip and whole genome sequencing in human population genetic analysis.BMC Genomics. 2018 Oct 29;19(1):778. doi: 10.1186/s12864-018-5168-x. BMC Genomics. 2018. PMID: 30373510 Free PMC article.
-
Methods and models for unravelling human evolutionary history.Nat Rev Genet. 2015 Dec;16(12):727-40. doi: 10.1038/nrg4005. Epub 2015 Nov 10. Nat Rev Genet. 2015. PMID: 26553329 Review.
-
Evolutionary genomics in Africa.Hum Mol Genet. 2021 Apr 26;30(R1):R1. doi: 10.1093/hmg/ddab030. Hum Mol Genet. 2021. PMID: 33899913 No abstract available.
-
Population genomics: a bridge from evolutionary history to genetic medicine.Hum Mol Genet. 2001 Oct 1;10(20):2199-207. doi: 10.1093/hmg/10.20.2199. Hum Mol Genet. 2001. PMID: 11673402 Review.
-
Constructing genomic maps of positive selection in humans: where do we go from here?Genome Res. 2009 May;19(5):711-22. doi: 10.1101/gr.086652.108. Genome Res. 2009. PMID: 19411596 Free PMC article.
Cited by
-
Recent explosive human population growth has resulted in an excess of rare genetic variants.Science. 2012 May 11;336(6082):740-3. doi: 10.1126/science.1217283. Science. 2012. PMID: 22582263 Free PMC article.
-
The role and challenges of exome sequencing in studies of human diseases.Front Genet. 2013 Aug 26;4:160. doi: 10.3389/fgene.2013.00160. Front Genet. 2013. PMID: 24032039 Free PMC article. Review.
-
Multiple recombination events between two cytochrome P450 loci contribute to global pyrethroid resistance in Helicoverpa armigera.PLoS One. 2018 Nov 1;13(11):e0197760. doi: 10.1371/journal.pone.0197760. eCollection 2018. PLoS One. 2018. PMID: 30383872 Free PMC article.
-
Efficient genome-wide genotyping strategies and data integration in crop plants.Theor Appl Genet. 2018 Mar;131(3):499-511. doi: 10.1007/s00122-018-3056-z. Epub 2018 Jan 19. Theor Appl Genet. 2018. PMID: 29352324 Review.
-
Personal and population genomics of human regulatory variation.Genome Res. 2012 Sep;22(9):1689-97. doi: 10.1101/gr.134890.111. Genome Res. 2012. PMID: 22955981 Free PMC article.
References
-
- Green RE, Krause J, Briggs AW, Maricic T, Stenzel U, Kircher M, Patterson N, Li H, Zhai W, Fritz MH, Hansen NF, Durand EY, Malaspinas AS, Jensen JD, Marques-Bonet T, Alkan C, Prüfer K, Meyer M, Burbano HA, Good JM, Schultz R, Aximu-Petri A, Butthof A, Höber B, Höffner B, Siegemund M, Weihmann A, Nusbaum C, Lander ES, Russ C. et al.A draft sequence of the Neandertal genome. Science. 2010;328:710–722. doi: 10.1126/science.1188021. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
