

Review
doi: 10.1186/gb-2011-12-9-227.
Computational and statistical approaches to analyzing variants identified by exome sequencing
Affiliations
- PMID: 21920052
- PMCID: PMC3308043
- DOI: 10.1186/gb-2011-12-9-227
Item in Clipboard
Review
Computational and statistical approaches to analyzing variants identified by exome sequencing
Genome Biol.
.
Abstract
New sequencing technology has enabled the identification of thousands of single nucleotide polymorphisms in the exome, and many computational and statistical approaches to identify disease-association signals have emerged.
Figures
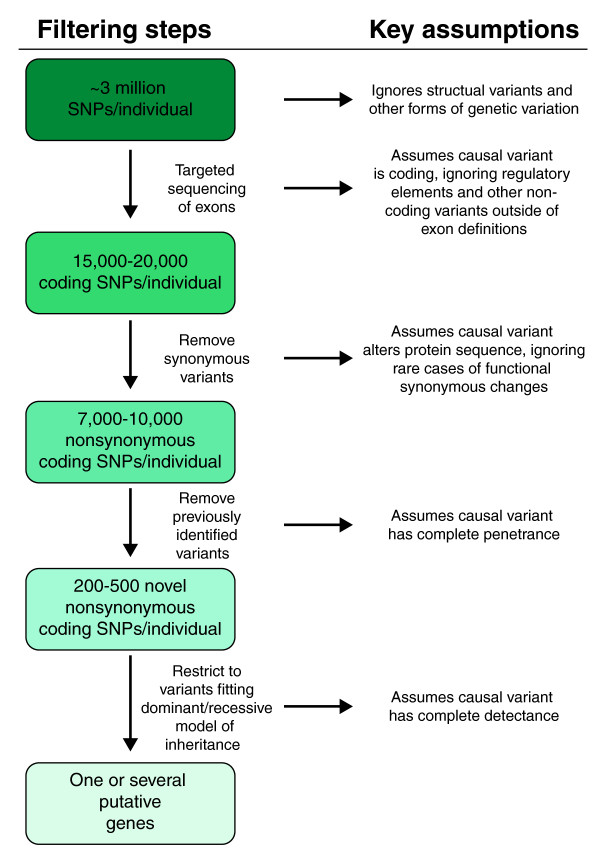
Typical heuristic filtering applied to exome sequencing projects aimed at novel gene discovery for Mendelian disorders, along with key assumptions at each step. Each individual carries approximately 3 million SNPs. Sequential filters shown here can be applied to reduce the number of potential disease-associated variants.
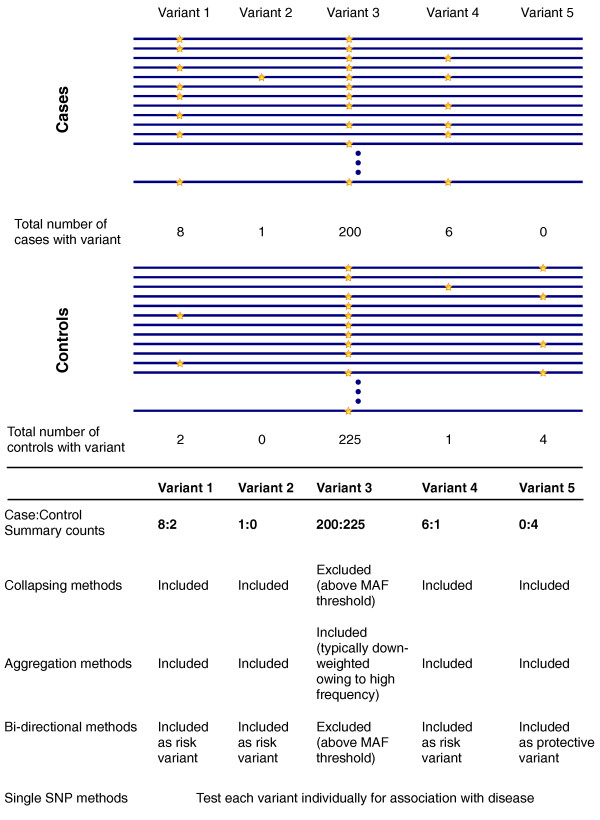
An illustration of rare variant association tests. Cases and controls from a hypothetical complex disease exome sequencing project are depicted. The horizontal bars indicate aligned exome sequences for individuals; stars indicate the presence of a non-reference allele. Variants 1 and 4 represent low-frequency variants with predominance in cases, Variant 2 represents a singleton, Variant 3 represents a common variant, and Variant 5 represents a low-frequency variant exclusive to controls. For simplicity, these variants are displayed with similar frequency, although very rare variants represent the majority of variation in real sequencing studies. As illustrated, the specific genetic architecture underlying the complex phenotype of interest is expected to have a large role in which test is most powerful for detecting an association. Collapsing methods may be best if a burden of rare variants drives the phenotype, whereas aggregation methods may be more powerful if the full allelic spectrum is contributory. Finally, for genes harboring both risk and protective alleles, bidirectional tests may be most appropriate. See Additional file 1 for examples of methods of each type. MAF, minor allele frequency.
References
-
- Online Mendelian Inheritance in Man. http://www.ncbi.nlm.nih.gov/omim/
-
- A Catalog of Published Genome-wide Association Studies. http://www.genome.gov/gwastudies/
-
- Worthey EA, Mayer AN, Syverson GD, Helbling D, Bonacci BB, Decker B, Serpe JM, Dasu T, Tschannen MR, Veith RL, Basehore MJ, Broeckel U, Tomita-Mitchell A, Arca MJ, Casper JT, Margolis DA, Bick DP, Hessner MJ, Routes JM, Verbsky JW, Jacob HJ, Dimmock DP. Making a definitive diagnosis: successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease. Genet Med. 2011;13:255–262. doi: 10.1097/GIM.0b013e3182088158. - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
