

Alcoholic liver disease: pathogenesis and new therapeutic targets
- PMID: 21920463
- PMCID: PMC3214974
- DOI: 10.1053/j.gastro.2011.09.002
Alcoholic liver disease: pathogenesis and new therapeutic targets
Abstract
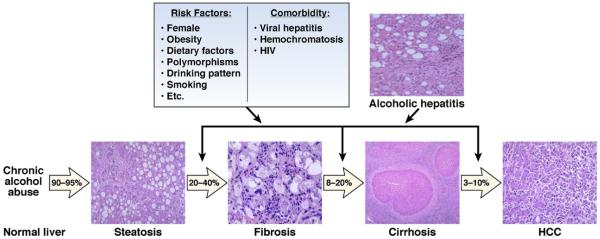
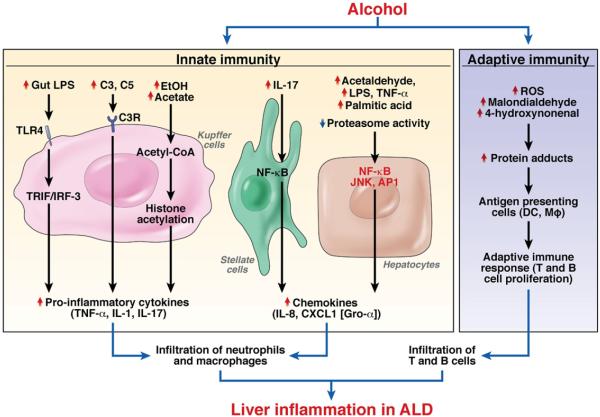
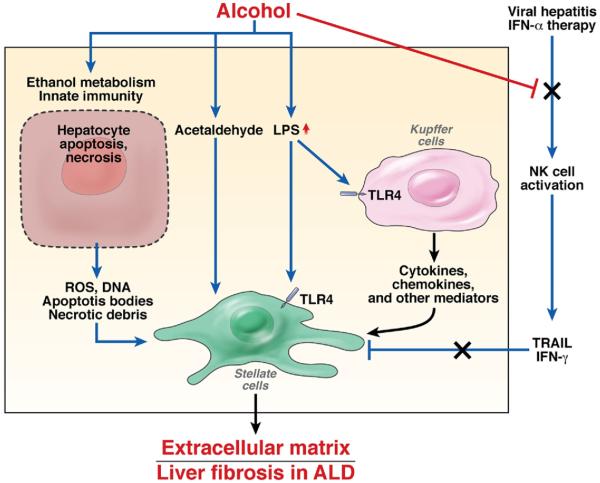
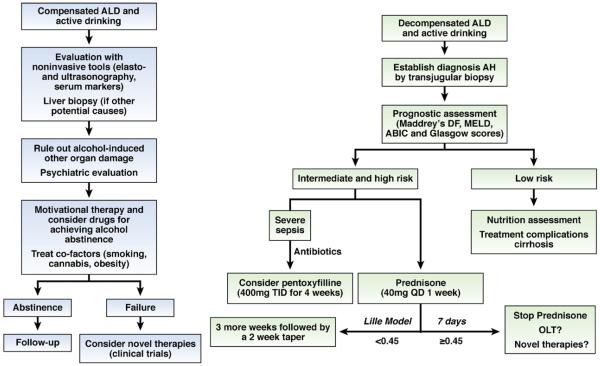
Alcoholic liver disease (ALD) is a major cause of chronic liver disease worldwide and can lead to fibrosis and cirrhosis. The latest surveillance report published by the National Institute on Alcohol Abuse and Alcoholism showed that liver cirrhosis was the 12th leading cause of death in the United States, with a total of 29,925 deaths in 2007, 48% of which were alcohol related. The spectrum of ALD includes simple steatosis, alcoholic hepatitis, fibrosis, cirrhosis, and superimposed hepatocellular carcinoma. Early work on the pathogenesis of the disease focused on ethanol metabolism-associated oxidative stress and glutathione depletion, abnormal methionine metabolism, malnutrition, and production of endotoxins that activate Kupffer cells. We review findings from recent studies that have characterized specific intracellular signaling pathways, transcriptional factors, aspects of innate immunity, chemokines, epigenetic features, microRNAs, and stem cells that are associated with ALD, improving our understanding of its pathogenesis. Despite this progress, no targeted therapies are available. The cornerstone of treatment for alcoholic hepatitis remains as it was 40 years ago: abstinence, nutritional support, and corticosteroids. There is an urgent need to develop new pathophysiology-oriented therapies. Recent translational studies of human samples and animal models have identified promising therapeutic targets.
Copyright © 2011 AGA Institute. Published by Elsevier Inc. All rights reserved.
Figures

References
-
- Rehm J, Mathers C, Popova S, et al. Global burden of disease and injury and economic cost attributable to alcohol use and alcohol-use disorders. Lancet. 2009;373:2223–2233. - PubMed
-
- Yoon Y, Yi H. Liver cirrhosis mortality in the United States, 1970-2007 surveillance report #88. NIAAA homepage. 2010 Available at: http://pubs.niaaa.nih.gov/publications/surveillance88/Cirr07.htm.
-
- Lieber CS. Susceptibility to alcohol-related liver injury. Alcohol Alcohol Suppl. 1994;2:315–326. - PubMed
-
- Thurman RG, Bradford BU, Iimuro Y, et al. The role of gut-derived bacterial toxins and free radicals in alcohol-induced liver injury. J Gastroenterol Hepatol. 1998;13(Suppl):S39–S50. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
