

Assessing and managing risk when sharing aggregate genetic variant data
- PMID: 21921928
- PMCID: PMC3349221
- DOI: 10.1038/nrg3067
Assessing and managing risk when sharing aggregate genetic variant data
Erratum in
- Nat Rev Genet. 2011 Nov;12(11):801
Abstract
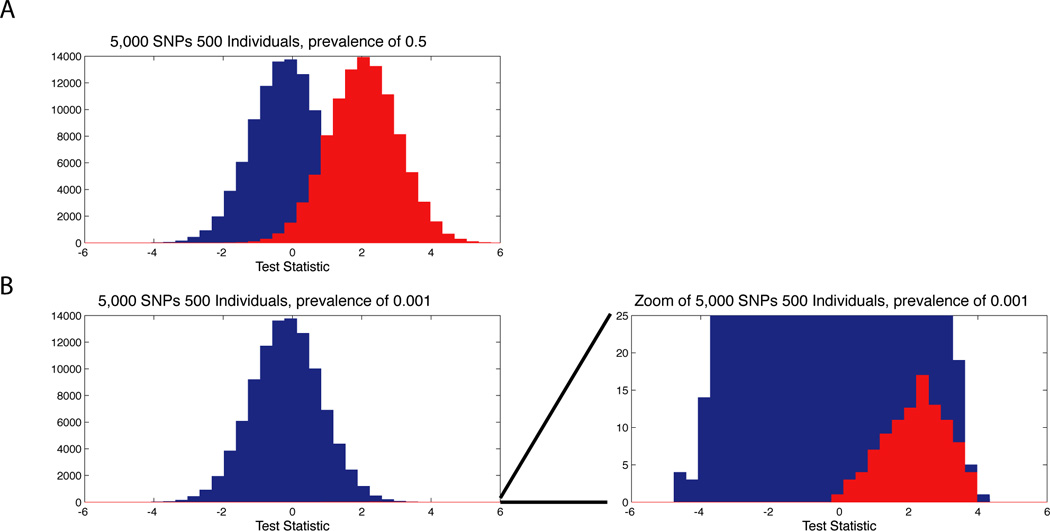
Access to genetic data across studies is an important aspect of identifying new genetic associations through genome-wide association studies (GWASs). Meta-analysis across multiple GWASs with combined cohort sizes of tens of thousands of individuals often uncovers many more genome-wide associated loci than the original individual studies; this emphasizes the importance of tools and mechanisms for data sharing. However, even sharing summary-level data, such as allele frequencies, inherently carries some degree of privacy risk to study participants. Here we discuss mechanisms and resources for sharing data from GWASs, particularly focusing on approaches for assessing and quantifying the privacy risks to participants that result from the sharing of summary-level data.
Figures
References
-
- Hirschhorn JN, Daly MJ. Genome-wide association studies for common diseases and complex traits. Nature reviews. Genetics. 2005;6:95–108. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
