

Using extreme phenotype sampling to identify the rare causal variants of quantitative traits in association studies
- PMID: 21922541
- PMCID: PMC4238184
- DOI: 10.1002/gepi.20628
Using extreme phenotype sampling to identify the rare causal variants of quantitative traits in association studies
Abstract
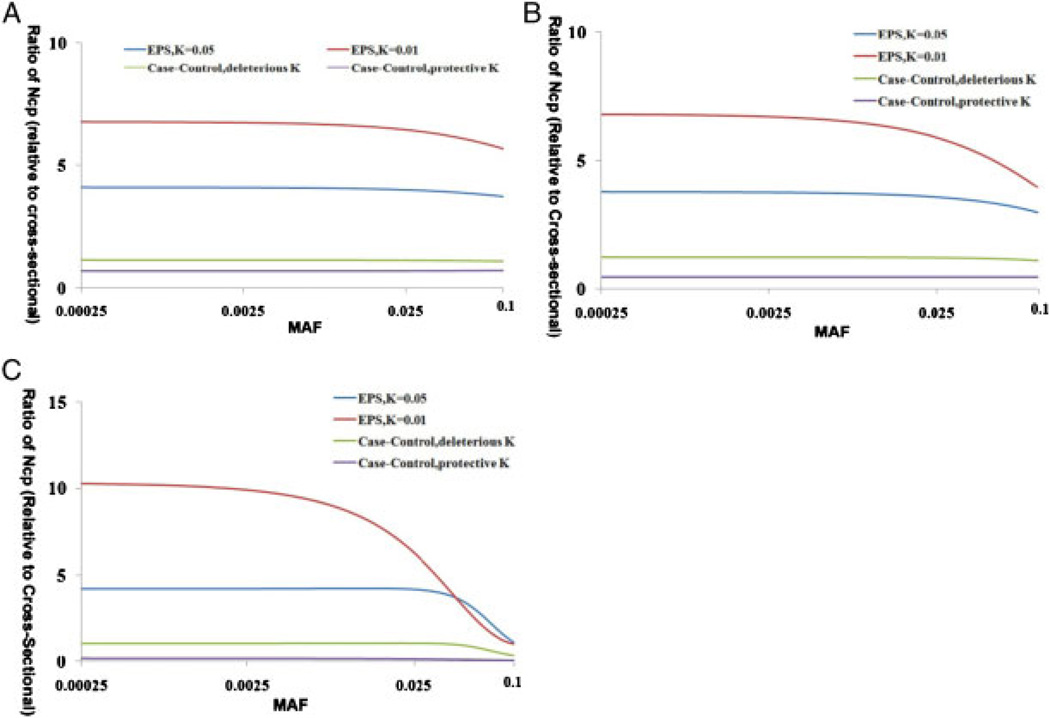
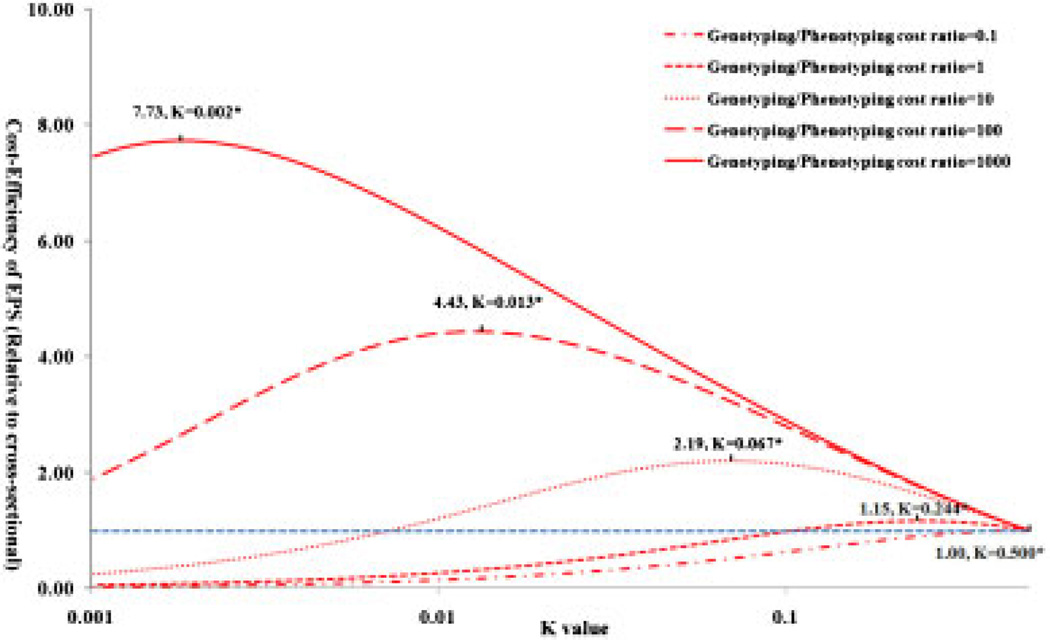
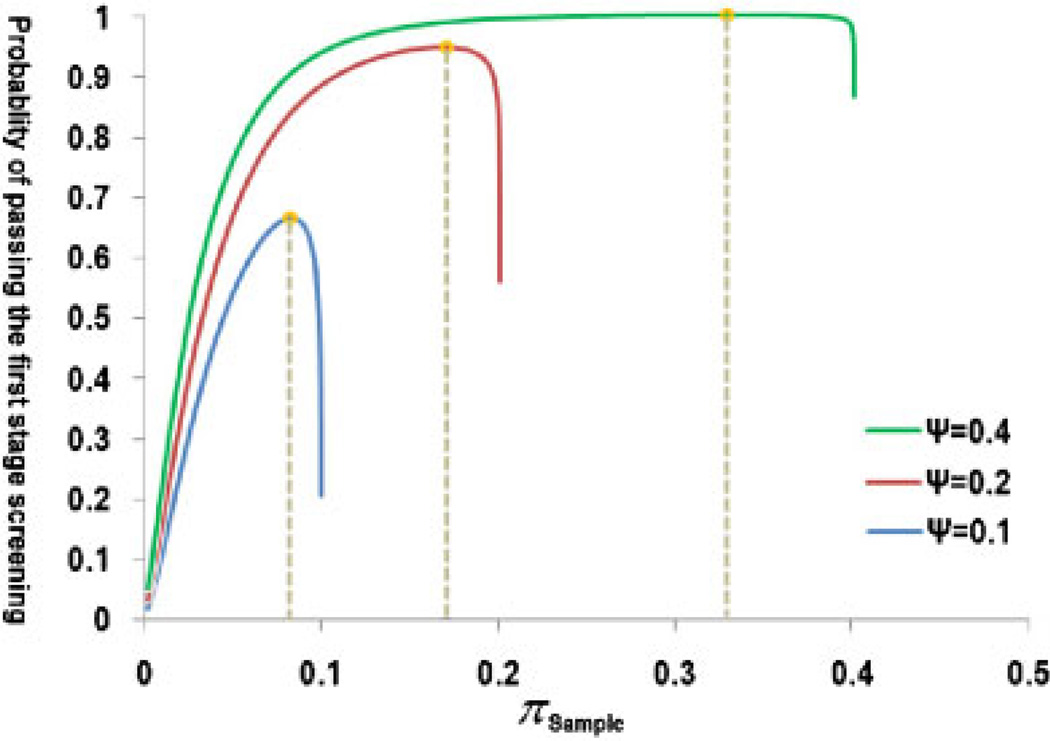
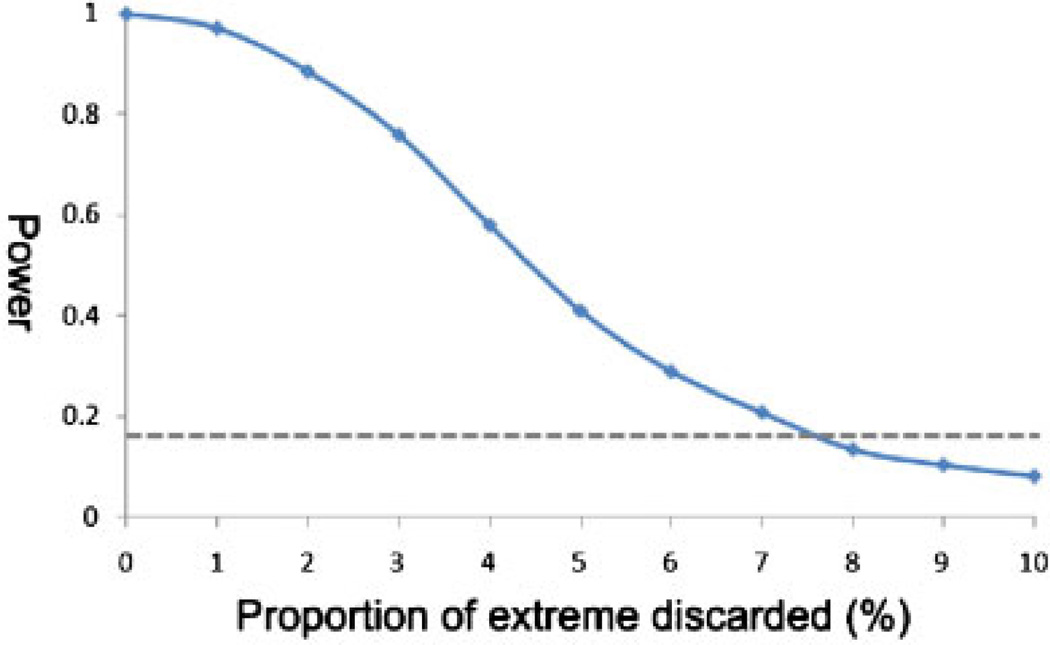
Variants identified in recent genome-wide association studies based on the common-disease common-variant hypothesis are far from fully explaining the hereditability of complex traits. Rare variants may, in part, explain some of the missing hereditability. Here, we explored the advantage of the extreme phenotype sampling in rare-variant analysis and refined this design framework for future large-scale association studies on quantitative traits. We first proposed a power calculation approach for a likelihood-based analysis method. We then used this approach to demonstrate the potential advantages of extreme phenotype sampling for rare variants. Next, we discussed how this design can influence future sequencing-based association studies from a cost-efficiency (with the phenotyping cost included) perspective. Moreover, we discussed the potential of a two-stage design with the extreme sample as the first stage and the remaining nonextreme subjects as the second stage. We demonstrated that this two-stage design is a cost-efficient alternative to the one-stage cross-sectional design or traditional two-stage design. We then discussed the analysis strategies for this extreme two-stage design and proposed a corresponding design optimization procedure. To address many practical concerns, for example measurement error or phenotypic heterogeneity at the very extremes, we examined an approach in which individuals with very extreme phenotypes are discarded. We demonstrated that even with a substantial proportion of these extreme individuals discarded, an extreme-based sampling can still be more efficient. Finally, we expanded the current analysis and design framework to accommodate the CMC approach where multiple rare-variants in the same gene region are analyzed jointly.
© 2011 Wiley Periodicals, Inc.
Figures
References
-
- Asimit J, Zeggini E. Rare variant association analysis methods for complex traits. Annu Rev Genet. 2010;44:293–308. - PubMed
-
- Chobanian AV, Bakris GI, Black HR, Cushman WC, Green LA, Izzo JL, Jr, Jones DW, Materson BJ, Oparil S, Wright JT, Rocella EJ. Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National Heart, Lung, and Blood Institute; National High Blood Pressure Education Program Coordinating Committee. 2003. Seventh report of the joint national committee on prevention, detection, evaluation, and treatment of high blood pressure. Hypertension. 2003;42:1206–1252. - PubMed
Publication types
MeSH terms
Grants and funding
- U01HG005927/HG/NHGRI NIH HHS/United States
- U01 ES015090/ES/NIEHS NIH HHS/United States
- CA084735/CA/NCI NIH HHS/United States
- R01 GM069890/GM/NIGMS NIH HHS/United States
- R01 ES016813/ES/NIEHS NIH HHS/United States
- P50 CA084735/CA/NCI NIH HHS/United States
- GM069890/GM/NIGMS NIH HHS/United States
- U01 DA020830/DA/NIDA NIH HHS/United States
- U19 CA148107/CA/NCI NIH HHS/United States
- R01 ES019876/ES/NIEHS NIH HHS/United States
- ES015090/ES/NIEHS NIH HHS/United States
- DA020830/DA/NIDA NIH HHS/United States
- U01 HG005927/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
