

Modeling the relative relationship of transcription factor binding and histone modifications to gene expression levels in mouse embryonic stem cells
- PMID: 21926158
- PMCID: PMC3258143
- DOI: 10.1093/nar/gkr752
Modeling the relative relationship of transcription factor binding and histone modifications to gene expression levels in mouse embryonic stem cells
Abstract
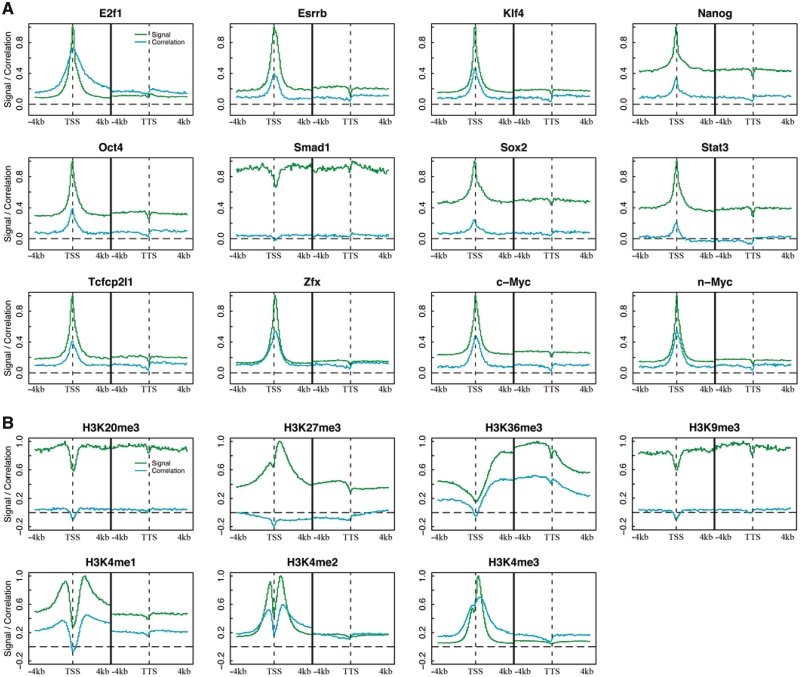
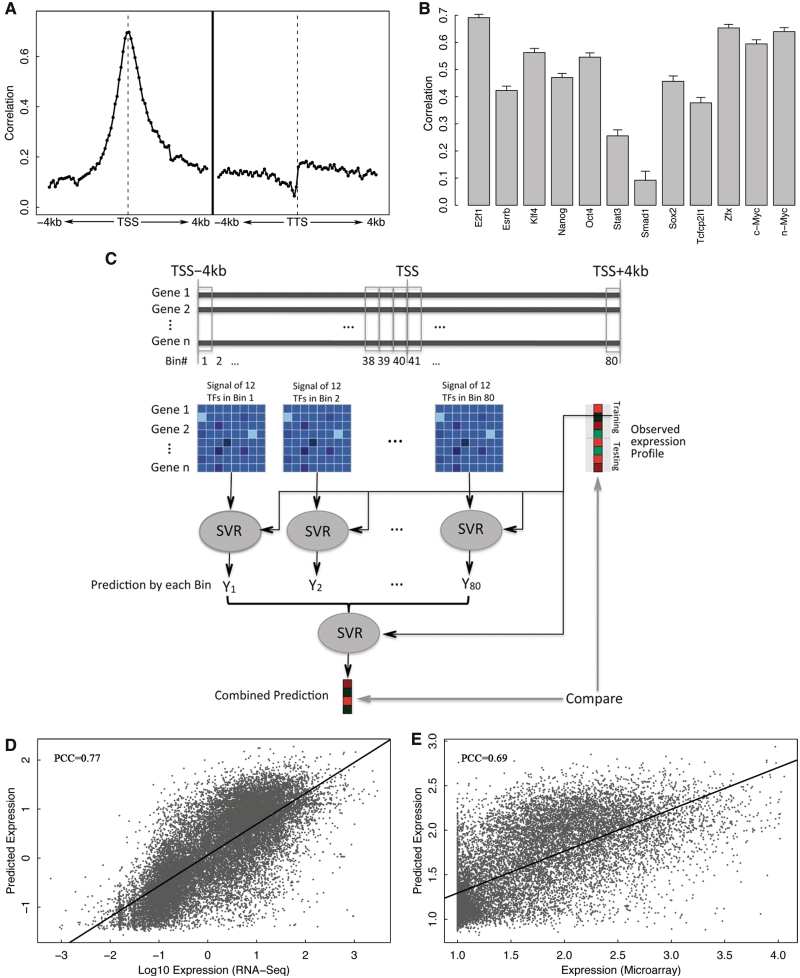
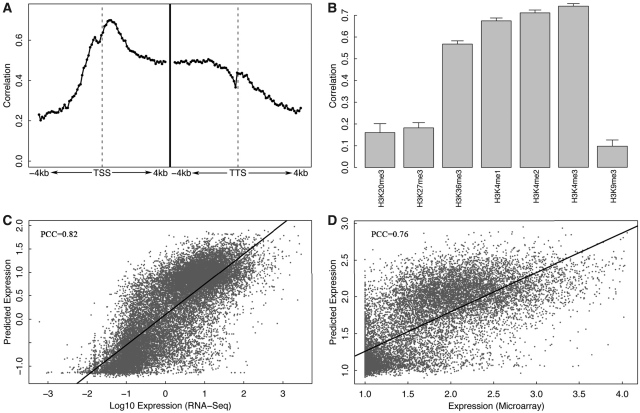
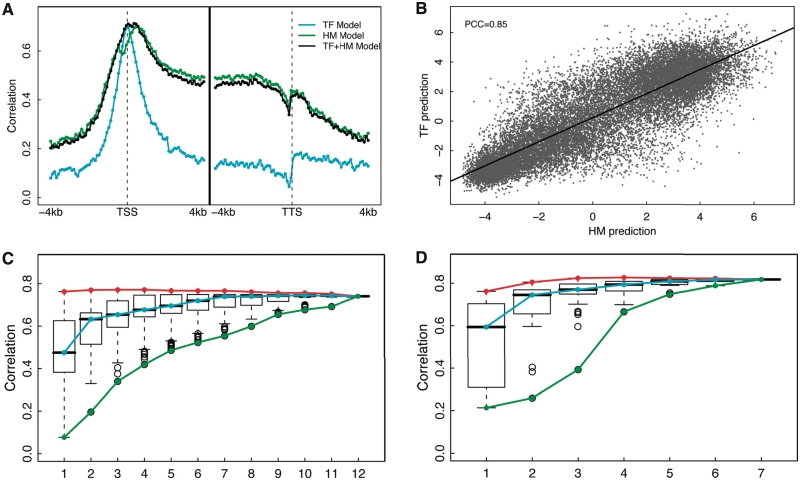
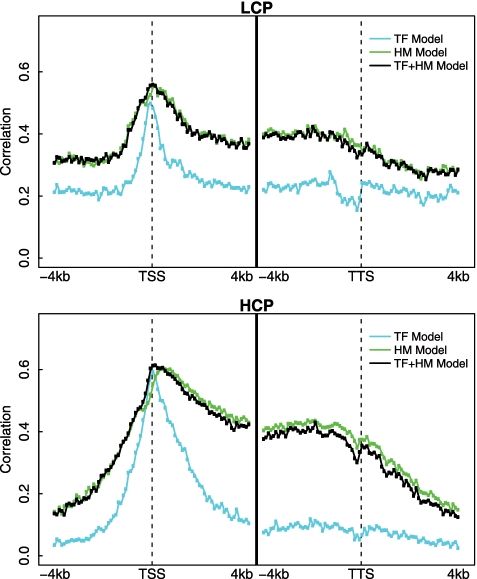
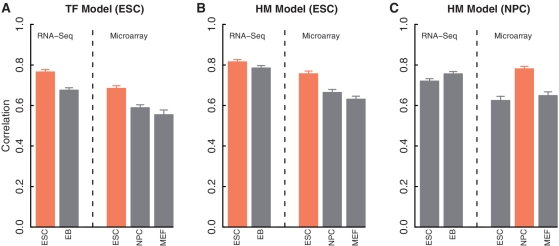
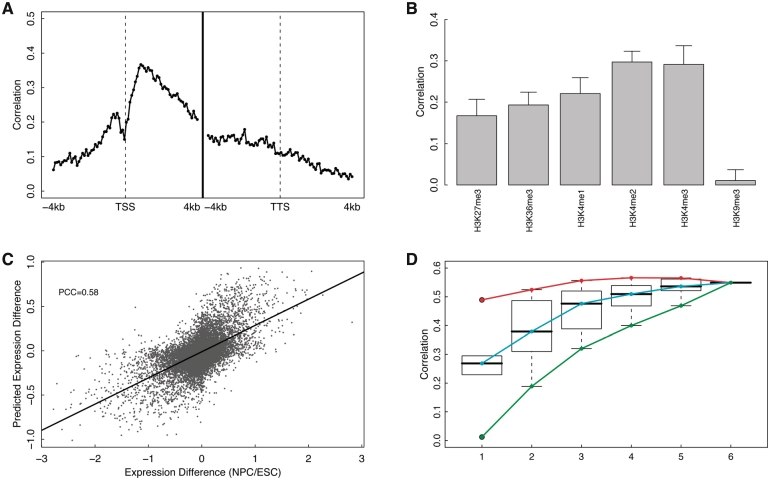
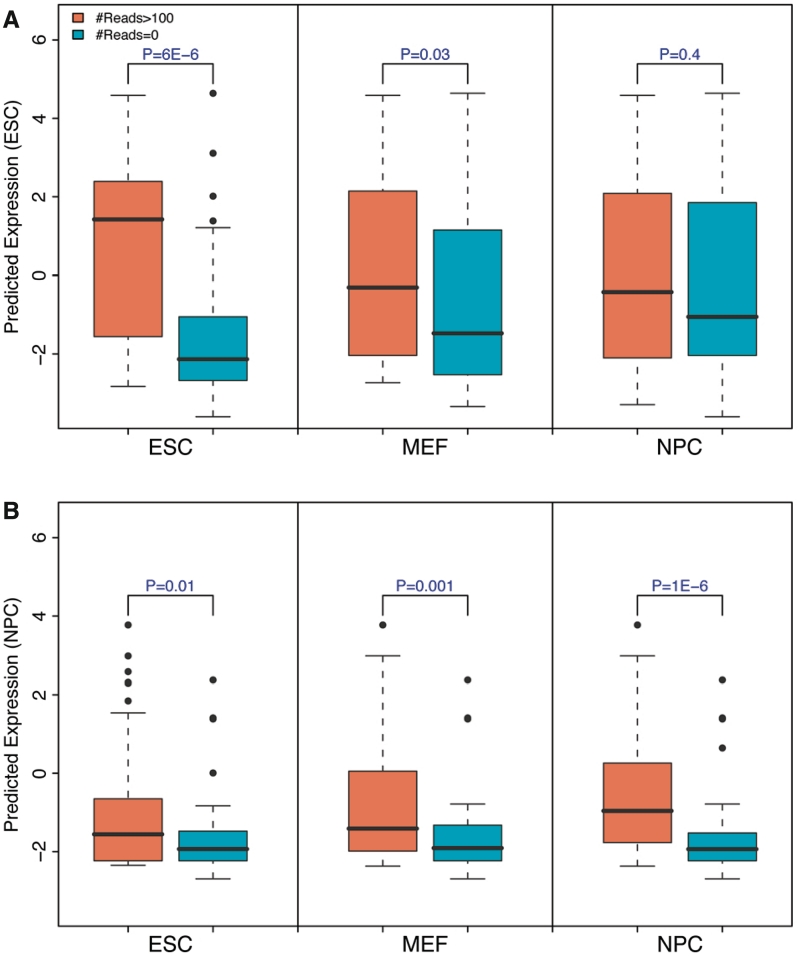
Transcription factor (TF) binding and histone modification (HM) are important for the precise control of gene expression. Hence, we constructed statistical models to relate these to gene expression levels in mouse embryonic stem cells. While both TF binding and HMs are highly 'predictive' of gene expression levels (in a statistical, but perhaps not strictly mechanistic, sense), we find they show distinct differences in the spatial patterning of their predictive strength: TF binding achieved the highest predictive power in a small DNA region centered at the transcription start sites of genes, while the HMs exhibited high predictive powers across a wide region around genes. Intriguingly, our results suggest that TF binding and HMs are redundant in strict statistical sense for predicting gene expression. We also show that our TF and HM models are cell line specific; specifically, TF binding and HM are more predictive of gene expression in the same cell line, and the differential gene expression between cell lines is predictable by differential HMs. Finally, we found that the models trained solely on protein-coding genes are predictive of expression levels of microRNAs, suggesting that their regulation by TFs and HMs may share a similar mechanism to that for protein-coding genes.
Figures
References
-
- Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–412. - PubMed
-
- Kurdistani SK, Tavazoie S, Grunstein M. Mapping global histone acetylation patterns to gene expression. Cell. 2004;117:721–733. - PubMed
-
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. - PubMed
-
- Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128:707–719. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
