

Efficient de novo assembly of single-cell bacterial genomes from short-read data sets
- PMID: 21926975
- PMCID: PMC3558281
- DOI: 10.1038/nbt.1966
Efficient de novo assembly of single-cell bacterial genomes from short-read data sets
Abstract
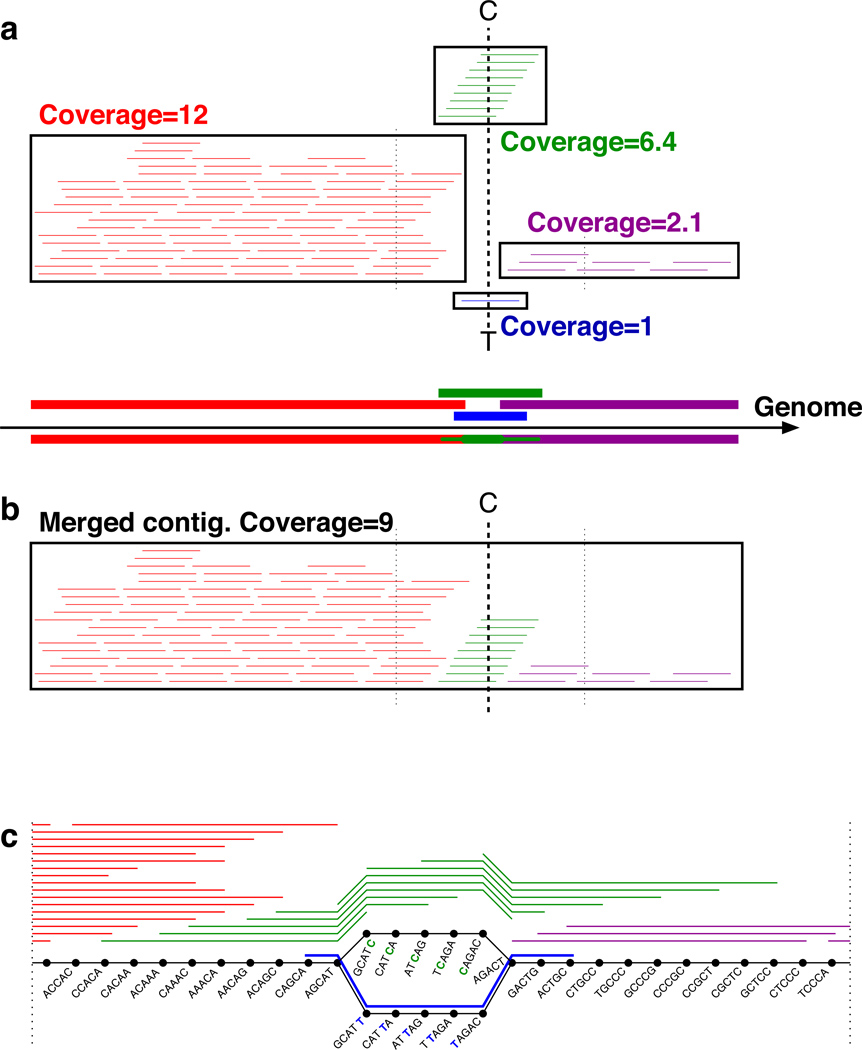
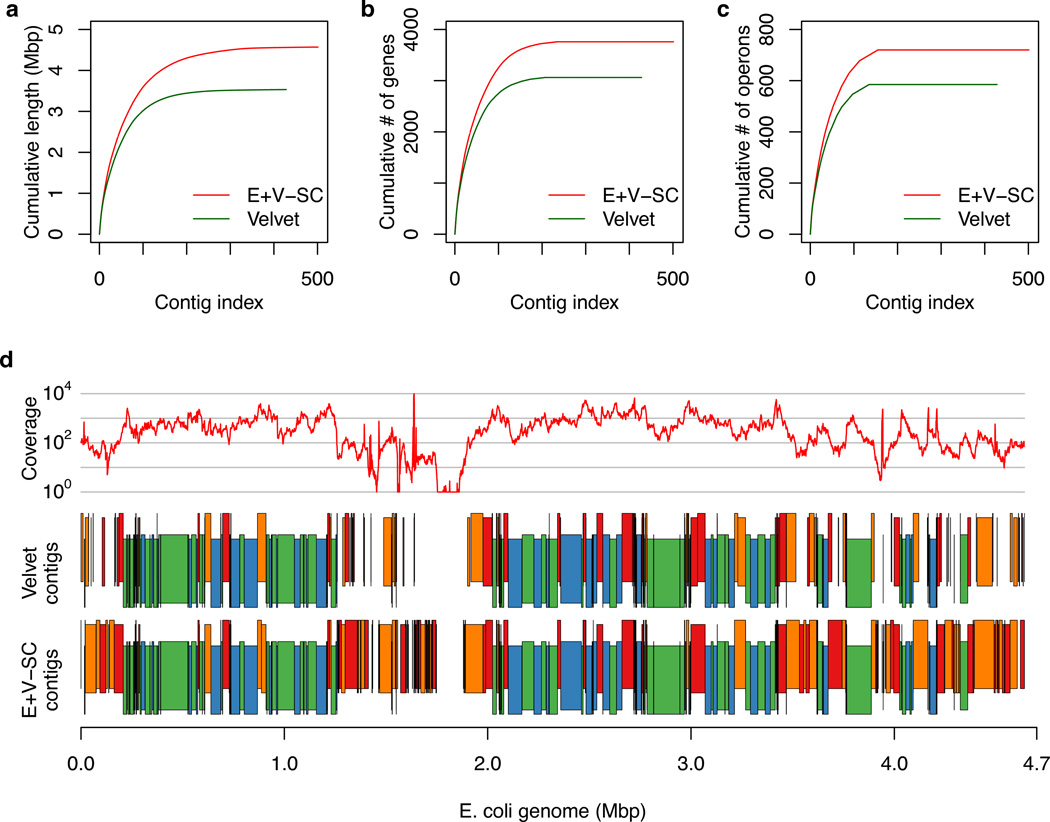
Whole genome amplification by the multiple displacement amplification (MDA) method allows sequencing of DNA from single cells of bacteria that cannot be cultured. Assembling a genome is challenging, however, because MDA generates highly nonuniform coverage of the genome. Here we describe an algorithm tailored for short-read data from single cells that improves assembly through the use of a progressively increasing coverage cutoff. Assembly of reads from single Escherichia coli and Staphylococcus aureus cells captures >91% of genes within contigs, approaching the 95% captured from an assembly based on many E. coli cells. We apply this method to assemble a genome from a single cell of an uncultivated SAR324 clade of Deltaproteobacteria, a cosmopolitan bacterial lineage in the global ocean. Metabolic reconstruction suggests that SAR324 is aerobic, motile and chemotaxic. Our approach enables acquisition of genome assemblies for individual uncultivated bacteria using only short reads, providing cell-specific genetic information absent from metagenomic studies.
Figures

Comment in
-
Picking up the pieces.Nat Methods. 2011 Nov;8(11):896-7. doi: 10.1038/nmeth.1753. Nat Methods. 2011. PMID: 22148156 No abstract available.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
