

Stress-stimulated mitogen-activated protein kinases control the stability and activity of the Cdt1 DNA replication licensing factor
- PMID: 21930785
- PMCID: PMC3209262
- DOI: 10.1128/MCB.06163-11
Stress-stimulated mitogen-activated protein kinases control the stability and activity of the Cdt1 DNA replication licensing factor
Abstract
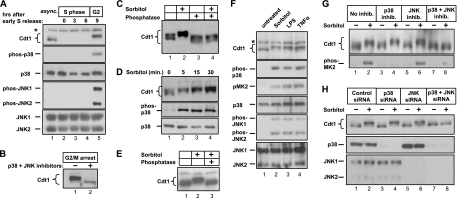
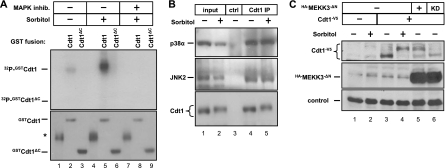
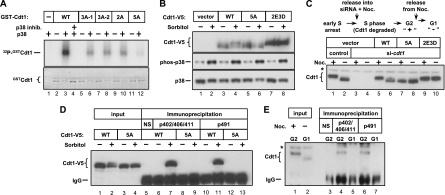
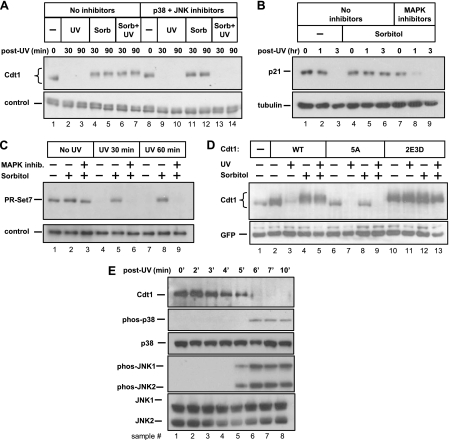
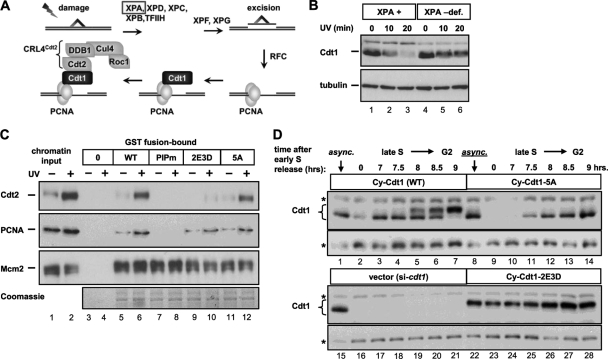
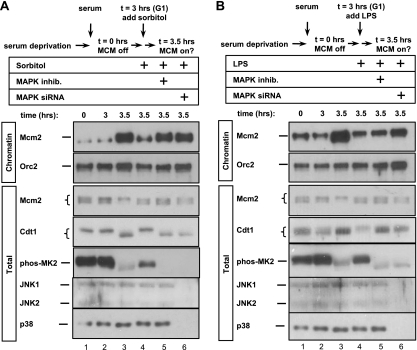
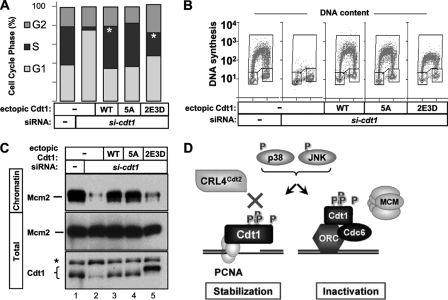
DNA replication is tightly coordinated both with cell cycle cues and with responses to extracellular signals to maintain genome stability. We discovered that human Cdt1, an essential origin licensing protein whose activity must be restricted to G(1) phase, is a substrate of the stress-activated mitogen-activated protein (MAP) kinases p38 and c-Jun N-terminal kinase (JNK). These MAP kinases phosphorylate Cdt1 both during unperturbed G(2) phase and during an acute stress response. Phosphorylation renders Cdt1 resistant to ubiquitin-mediated degradation during S phase and after DNA damage by blocking Cdt1 binding to the Cul4 adaptor, Cdt2. Mutations that block normal cell cycle-regulated MAP kinase-mediated phosphorylation interfere with rapid Cdt1 reaccumulation at the end of S phase. Phosphomimetic mutations recapitulate the stabilizing effects of Cdt1 phosphorylation but also reduce the ability of Cdt1 to support origin licensing. Two other CRL4(Cdt2) targets, the cyclin-dependent kinase (CDK) inhibitor p21 and the methyltransferase PR-Set7/Set8, are similarly stabilized by MAP kinase activity. These findings support a model in which MAP kinase activity in G(2) promotes reaccumulation of a low-activity Cdt1 isoform after replication is complete.
Figures
Similar articles
-
CDK1-dependent inhibition of the E3 ubiquitin ligase CRL4CDT2 ensures robust transition from S Phase to Mitosis.J Biol Chem. 2015 Jan 2;290(1):556-67. doi: 10.1074/jbc.M114.614701. Epub 2014 Nov 19. J Biol Chem. 2015. PMID: 25411249 Free PMC article.
-
Selective ubiquitylation of p21 and Cdt1 by UBCH8 and UBE2G ubiquitin-conjugating enzymes via the CRL4Cdt2 ubiquitin ligase complex.Mol Cell Biol. 2011 Aug;31(15):3136-45. doi: 10.1128/MCB.05496-11. Epub 2011 May 31. Mol Cell Biol. 2011. PMID: 21628527 Free PMC article.
-
Checkpoint kinase ATR phosphorylates Cdt2, a substrate receptor of CRL4 ubiquitin ligase, and promotes the degradation of Cdt1 following UV irradiation.PLoS One. 2012;7(9):e46480. doi: 10.1371/journal.pone.0046480. Epub 2012 Sep 28. PLoS One. 2012. PMID: 23029527 Free PMC article.
-
Regulation of DNA Replication Licensing and Re-Replication by Cdt1.Int J Mol Sci. 2021 May 14;22(10):5195. doi: 10.3390/ijms22105195. Int J Mol Sci. 2021. PMID: 34068957 Free PMC article. Review.
-
CRL4Cdt2 Ubiquitin Ligase, A Genome Caretaker Controlled by Cdt2 Binding to PCNA and DNA.Genes (Basel). 2022 Jan 29;13(2):266. doi: 10.3390/genes13020266. Genes (Basel). 2022. PMID: 35205311 Free PMC article. Review.
Cited by
-
SCF-FBXO31 E3 ligase targets DNA replication factor Cdt1 for proteolysis in the G2 phase of cell cycle to prevent re-replication.J Biol Chem. 2014 Jun 27;289(26):18514-25. doi: 10.1074/jbc.M114.559930. Epub 2014 May 14. J Biol Chem. 2014. PMID: 24828503 Free PMC article.
-
The p57 CDKi integrates stress signals into cell-cycle progression to promote cell survival upon stress.EMBO J. 2012 Jun 29;31(13):2952-64. doi: 10.1038/emboj.2012.122. Epub 2012 May 8. EMBO J. 2012. PMID: 22569127 Free PMC article.
-
Preparation for DNA replication: the key to a successful S phase.FEBS Lett. 2019 Oct;593(20):2853-2867. doi: 10.1002/1873-3468.13619. Epub 2019 Oct 15. FEBS Lett. 2019. PMID: 31556113 Free PMC article. Review.
-
Geminin deploys multiple mechanisms to regulate Cdt1 before cell division thus ensuring the proper execution of DNA replication.Proc Natl Acad Sci U S A. 2013 Jul 23;110(30):E2848-53. doi: 10.1073/pnas.1310677110. Epub 2013 Jul 8. Proc Natl Acad Sci U S A. 2013. PMID: 23836640 Free PMC article.
-
CDK1-dependent inhibition of the E3 ubiquitin ligase CRL4CDT2 ensures robust transition from S Phase to Mitosis.J Biol Chem. 2015 Jan 2;290(1):556-67. doi: 10.1074/jbc.M114.614701. Epub 2014 Nov 19. J Biol Chem. 2015. PMID: 25411249 Free PMC article.
References
-
- Aboussekhra A., et al. 1995. Mammalian DNA nucleotide excision repair reconstituted with purified protein components. Cell 80:859–868 - PubMed
-
- Alvarez E., et al. 1991. Pro-Leu-Ser/Thr-Pro is a consensus primary sequence for substrate protein phosphorylation. Characterization of the phosphorylation of c-myc and c-jun proteins by an epidermal growth factor receptor threonine 669 protein kinase. J. Biol. Chem. 266:15277–15285 - PubMed
-
- Arentson E., et al. 2002. Oncogenic potential of the DNA replication licensing protein CDT1. Oncogene 21:1150–1158 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous