

Binding of superantigen toxins into the CD28 homodimer interface is essential for induction of cytokine genes that mediate lethal shock
- PMID: 21931534
- PMCID: PMC3172200
- DOI: 10.1371/journal.pbio.1001149
Binding of superantigen toxins into the CD28 homodimer interface is essential for induction of cytokine genes that mediate lethal shock
Erratum in
-
Correction: Binding of Superantigen Toxins into the CD28 Homodimer Interface Is Essential for Induction of Cytokine Genes That Mediate Lethal Shock.PLoS Biol. 2015 Aug 21;13(8):e1002237. doi: 10.1371/journal.pbio.1002237. eCollection 2015 Aug. PLoS Biol. 2015. PMID: 26296256 Free PMC article. No abstract available.
Abstract
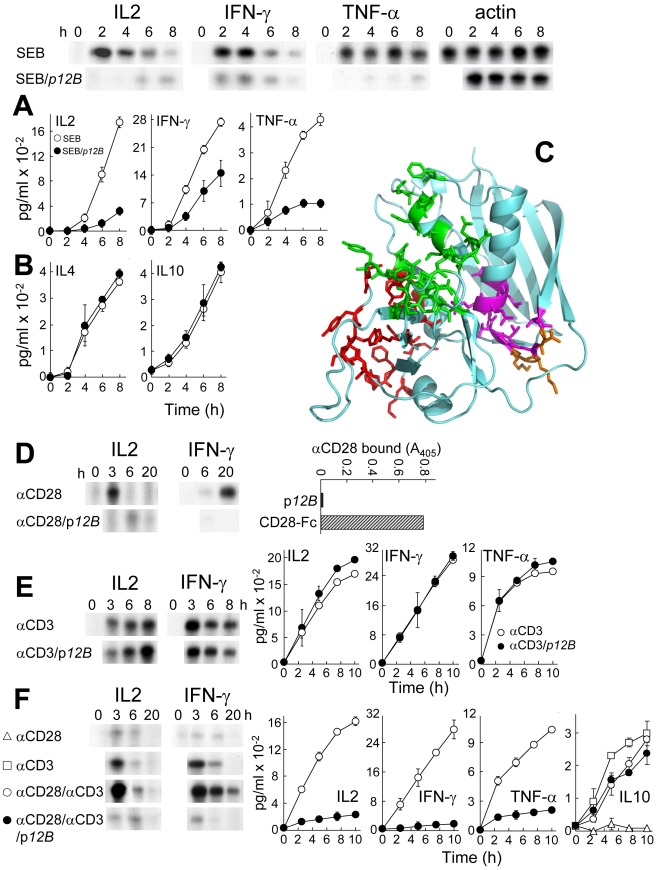
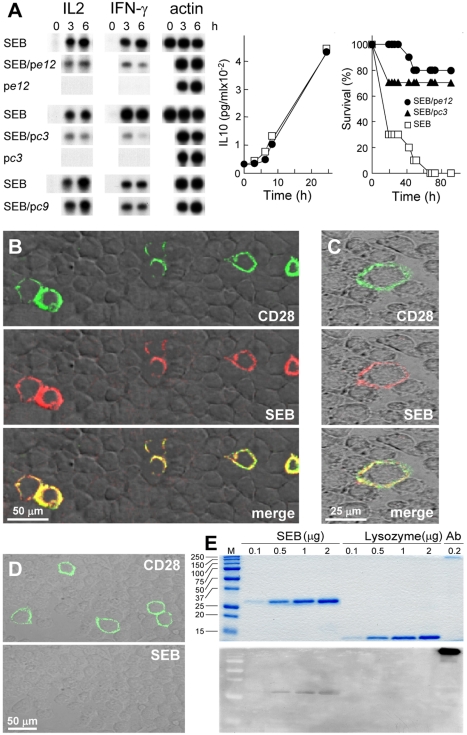
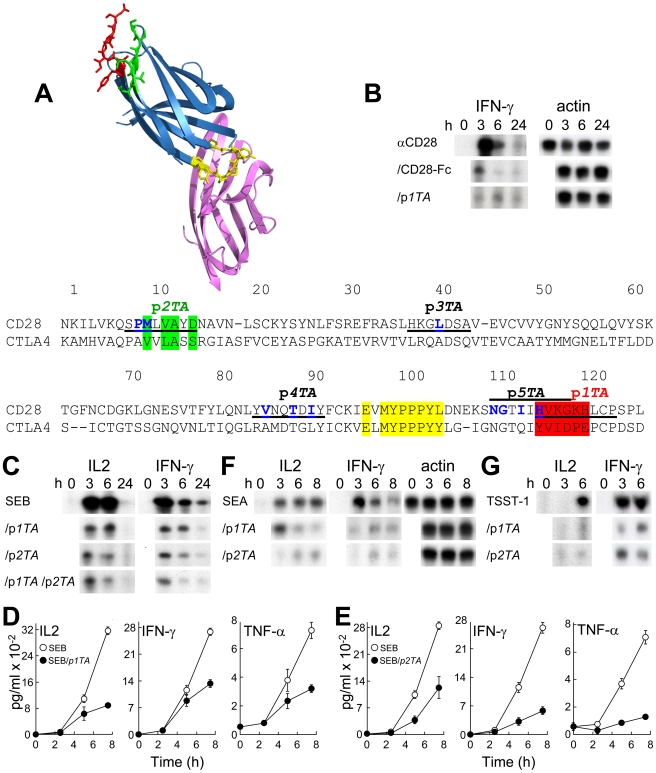
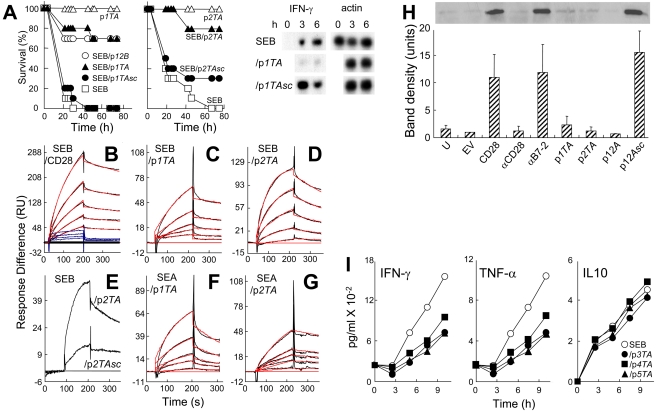
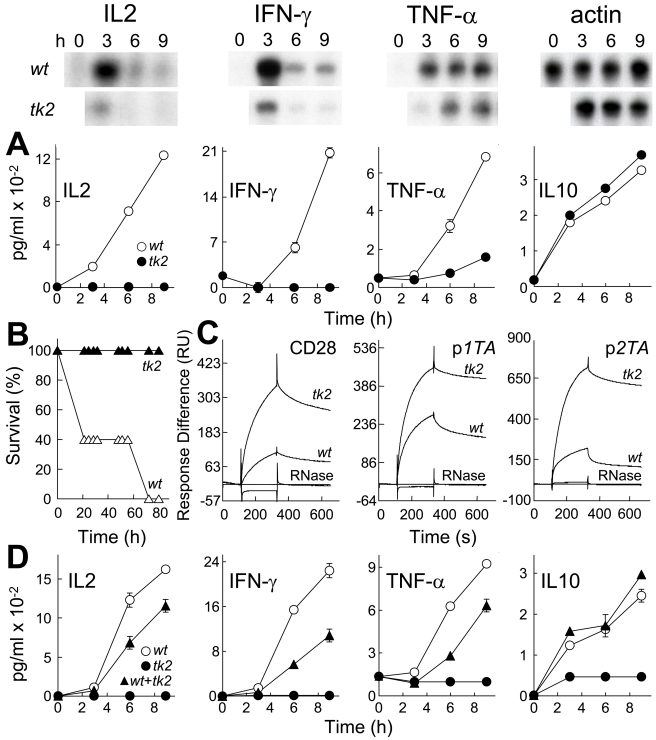
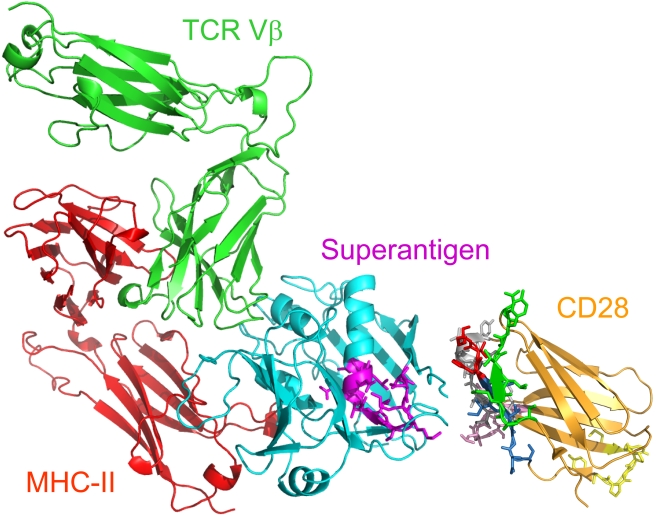
Bacterial superantigens, a diverse family of toxins, induce an inflammatory cytokine storm that can lead to lethal shock. CD28 is a homodimer expressed on T cells that functions as the principal costimulatory ligand in the immune response through an interaction with its B7 coligands, yet we show here that to elicit inflammatory cytokine gene expression and toxicity, superantigens must bind directly into the dimer interface of CD28. Preventing access of the superantigen to CD28 suffices to block its lethality. Mice were protected from lethal superantigen challenge by short peptide mimetics of the CD28 dimer interface and by peptides selected to compete with the superantigen for its binding site in CD28. Superantigens use a conserved β-strand/hinge/α-helix domain of hitherto unknown function to engage CD28. Mutation of this superantigen domain abolished inflammatory cytokine gene induction and lethality. Structural analysis showed that when a superantigen binds to the T cell receptor on the T cell and major histocompatibility class II molecule on the antigen-presenting cell, CD28 can be accommodated readily as third superantigen receptor in the quaternary complex, with the CD28 dimer interface oriented towards the β-strand/hinge/α-helix domain in the superantigen. Our findings identify the CD28 homodimer interface as a critical receptor target for superantigens. The novel role of CD28 as receptor for a class of microbial pathogens, the superantigen toxins, broadens the scope of pathogen recognition mechanisms.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
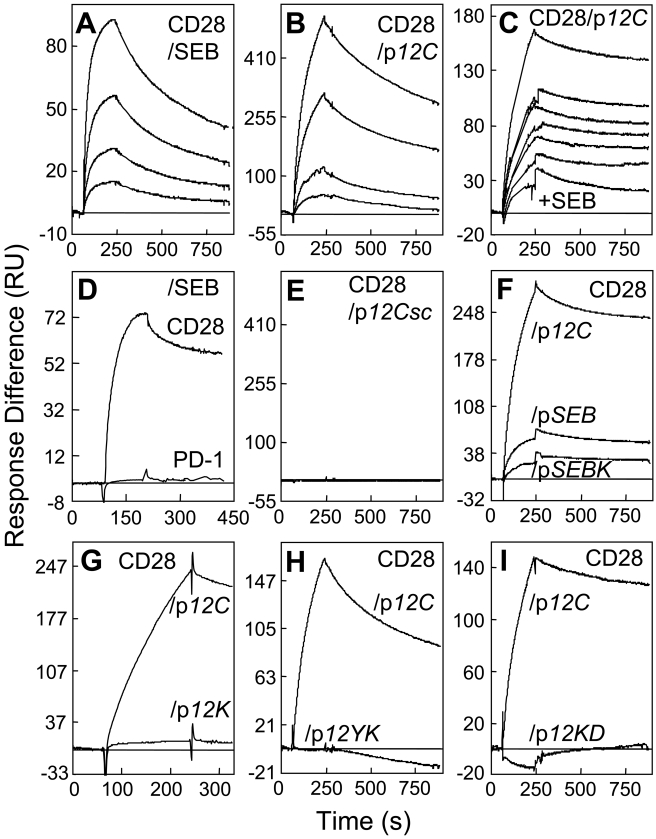
References
-
- Schwartz J. C, Zhang X, Fedorov A. A, Nathenson S. G, Almo S. C. Structural basis for co-stimulation by the human CTLA-4/B7-2 complex. Nature. 2001;410:604–608. - PubMed
-
- Sharpe A. H, Freeman G. J. The B7-CD28 superfamily. Nat Rev Immunol. 2002;2:116–126. - PubMed
-
- Riley J. L, June C. H. The CD28 family: a T-cell rheostat for therapeutic control of T-cell activation. Blood. 2005;105:13–21. - PubMed
-
- Lindsten T, Lee K. P, Harris E. S, Petryniak B, Craighead N, et al. Characterization of CTLA-4 structure and expression on human T cells. J Immunol. 1993;151:3489–3499. - PubMed
-
- Collins A. V, Brodie D. W, Gilbert R. J, Iaboni A, Manso-Sancho R, et al. The interaction properties of costimulatory molecules revisited. Immunity. 2002;17:201–210. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
