

Efficacy of synaptic inhibition depends on multiple, dynamically interacting mechanisms implicated in chloride homeostasis
- PMID: 21931544
- PMCID: PMC3169517
- DOI: 10.1371/journal.pcbi.1002149
Efficacy of synaptic inhibition depends on multiple, dynamically interacting mechanisms implicated in chloride homeostasis
Abstract
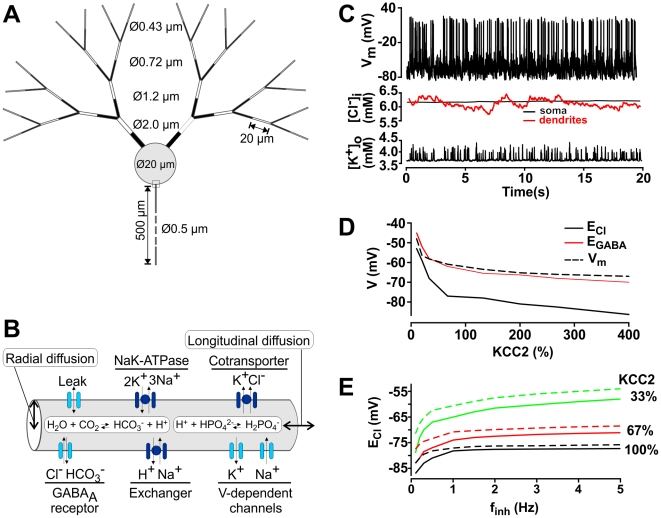
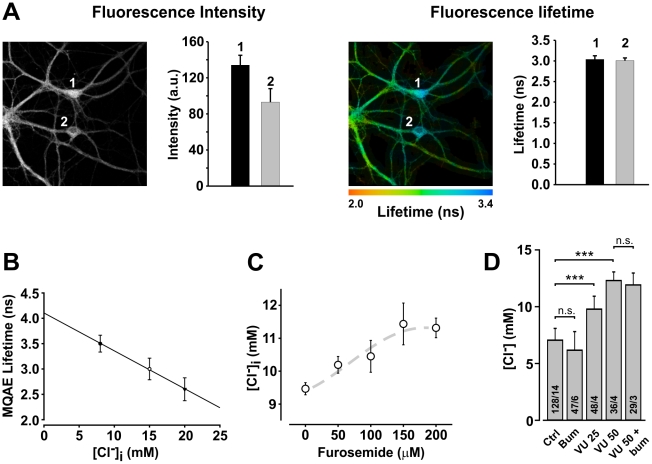
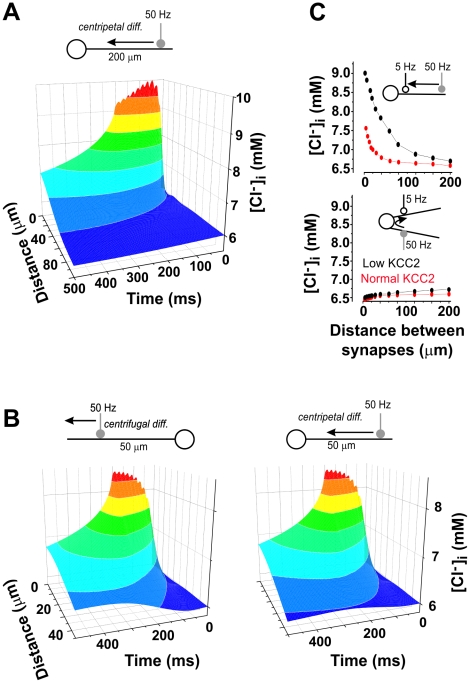
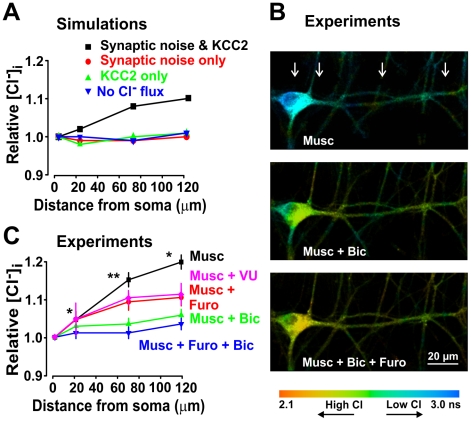
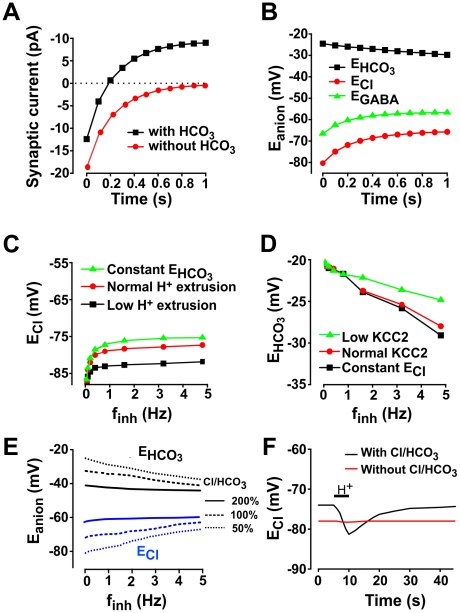
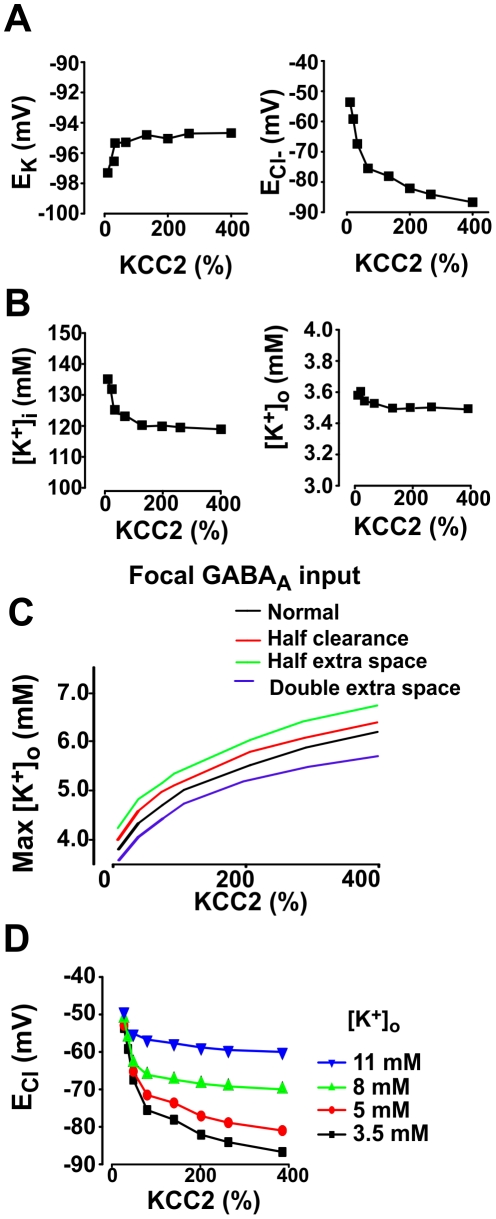
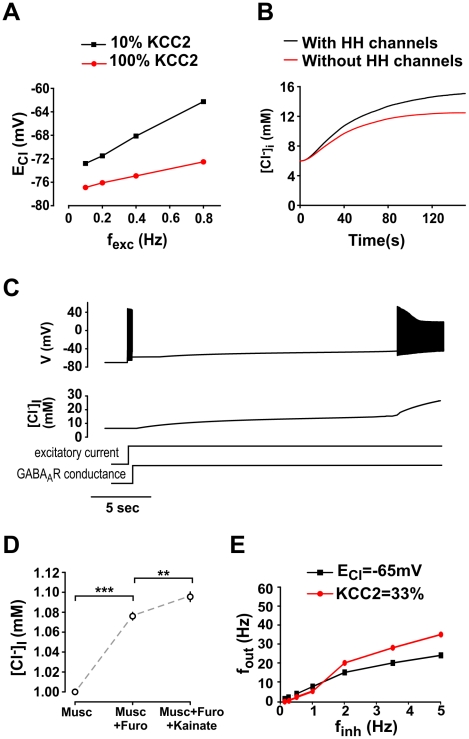
Chloride homeostasis is a critical determinant of the strength and robustness of inhibition mediated by GABA(A) receptors (GABA(A)Rs). The impact of changes in steady state Cl(-) gradient is relatively straightforward to understand, but how dynamic interplay between Cl(-) influx, diffusion, extrusion and interaction with other ion species affects synaptic signaling remains uncertain. Here we used electrodiffusion modeling to investigate the nonlinear interactions between these processes. Results demonstrate that diffusion is crucial for redistributing intracellular Cl(-) load on a fast time scale, whereas Cl(-)extrusion controls steady state levels. Interaction between diffusion and extrusion can result in a somato-dendritic Cl(-) gradient even when KCC2 is distributed uniformly across the cell. Reducing KCC2 activity led to decreased efficacy of GABA(A)R-mediated inhibition, but increasing GABA(A)R input failed to fully compensate for this form of disinhibition because of activity-dependent accumulation of Cl(-). Furthermore, if spiking persisted despite the presence of GABA(A)R input, Cl(-) accumulation became accelerated because of the large Cl(-) driving force that occurs during spikes. The resulting positive feedback loop caused catastrophic failure of inhibition. Simulations also revealed other feedback loops, such as competition between Cl(-) and pH regulation. Several model predictions were tested and confirmed by [Cl(-)](i) imaging experiments. Our study has thus uncovered how Cl(-) regulation depends on a multiplicity of dynamically interacting mechanisms. Furthermore, the model revealed that enhancing KCC2 activity beyond normal levels did not negatively impact firing frequency or cause overt extracellular K(-) accumulation, demonstrating that enhancing KCC2 activity is a valid strategy for therapeutic intervention.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Cell type-specific differences in chloride-regulatory mechanisms and GABA(A) receptor-mediated inhibition in rat substantia nigra.J Neurosci. 2003 Sep 10;23(23):8237-46. doi: 10.1523/JNEUROSCI.23-23-08237.2003. J Neurosci. 2003. PMID: 12967985 Free PMC article.
-
Could tuning of the inhibitory tone involve graded changes in neuronal chloride transport?Neuropharmacology. 2015 Aug;95:321-31. doi: 10.1016/j.neuropharm.2015.03.026. Epub 2015 Apr 3. Neuropharmacology. 2015. PMID: 25843644
-
GABAA receptor dependent synaptic inhibition rapidly tunes KCC2 activity via the Cl--sensitive WNK1 kinase.Nat Commun. 2017 Nov 24;8(1):1776. doi: 10.1038/s41467-017-01749-0. Nat Commun. 2017. PMID: 29176664 Free PMC article.
-
KCC2 membrane diffusion tunes neuronal chloride homeostasis.Neuropharmacology. 2020 Jun 1;169:107571. doi: 10.1016/j.neuropharm.2019.03.014. Epub 2019 Mar 11. Neuropharmacology. 2020. PMID: 30871970 Review.
-
Double-edged GABAergic synaptic transmission in seizures: The importance of chloride plasticity.Brain Res. 2018 Dec 15;1701:126-136. doi: 10.1016/j.brainres.2018.09.008. Epub 2018 Sep 7. Brain Res. 2018. PMID: 30201259 Review.
Cited by
-
Microglia disrupt mesolimbic reward circuitry in chronic pain.J Neurosci. 2015 Jun 3;35(22):8442-50. doi: 10.1523/JNEUROSCI.4036-14.2015. J Neurosci. 2015. PMID: 26041913 Free PMC article.
-
The effects of chloride dynamics on substantia nigra pars reticulata responses to pallidal and striatal inputs.Elife. 2020 Sep 7;9:e55592. doi: 10.7554/eLife.55592. Elife. 2020. PMID: 32894224 Free PMC article.
-
Microglia control neuronal network excitability via BDNF signalling.Neural Plast. 2013;2013:429815. doi: 10.1155/2013/429815. Epub 2013 Sep 5. Neural Plast. 2013. PMID: 24089642 Free PMC article. Review.
-
Mild KCC2 Hypofunction Causes Inconspicuous Chloride Dysregulation that Degrades Neural Coding.Front Cell Neurosci. 2016 Jan 29;9:516. doi: 10.3389/fncel.2015.00516. eCollection 2015. Front Cell Neurosci. 2016. PMID: 26858607 Free PMC article.
-
Biophysical models reveal the relative importance of transporter proteins and impermeant anions in chloride homeostasis.Elife. 2018 Sep 27;7:e39575. doi: 10.7554/eLife.39575. Elife. 2018. PMID: 30260315 Free PMC article.
References
-
- Hausser M, Major G, Stuart GJ. Differential shunting of EPSPs by action potentials. Science. 2001;291:138–141. - PubMed
-
- Staley KJ, Soldo BL, Proctor WR. Ionic mechanisms of neuronal excitation by inhibitory GABAA receptors. Science. 1995;269:977–981. - PubMed
-
- Kaila K, Voipio J. Postsynaptic Fall in intracellular pH induced by GABA-activated bicarbonate Cconductance. Nature. 1987;330:163–165. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
