

Novel molecular and computational methods improve the accuracy of insertion site analysis in Sleeping Beauty-induced tumors
- PMID: 21931803
- PMCID: PMC3172244
- DOI: 10.1371/journal.pone.0024668
Novel molecular and computational methods improve the accuracy of insertion site analysis in Sleeping Beauty-induced tumors
Abstract
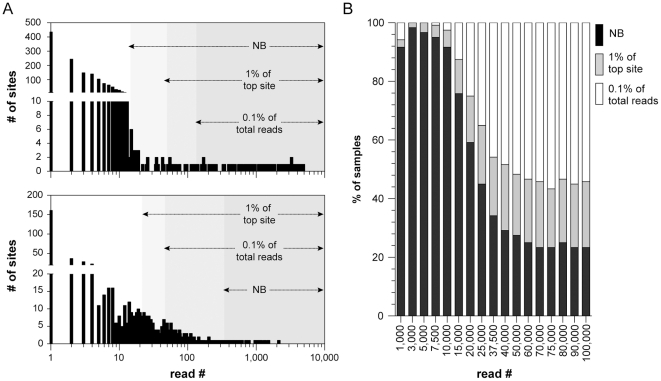
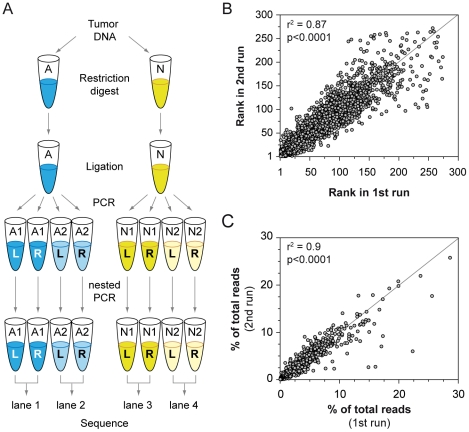
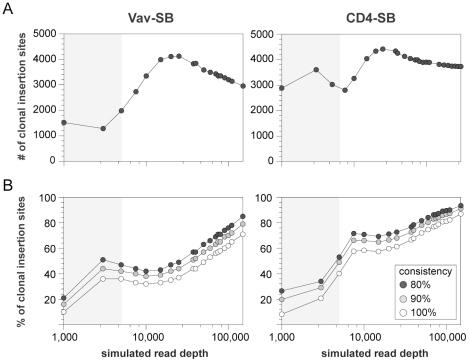
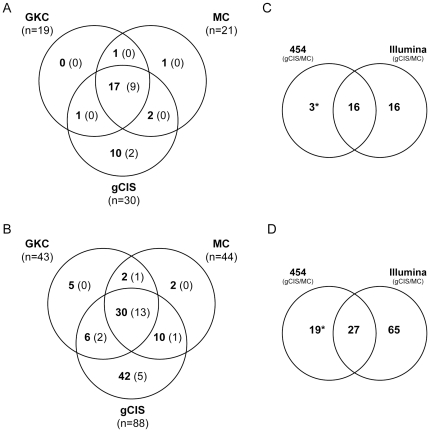
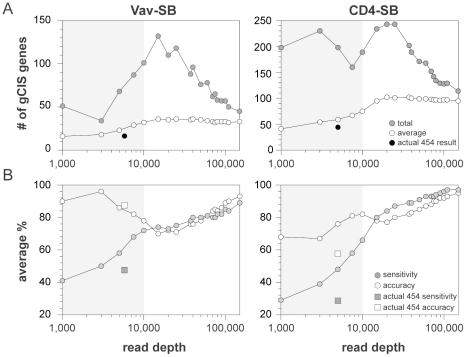
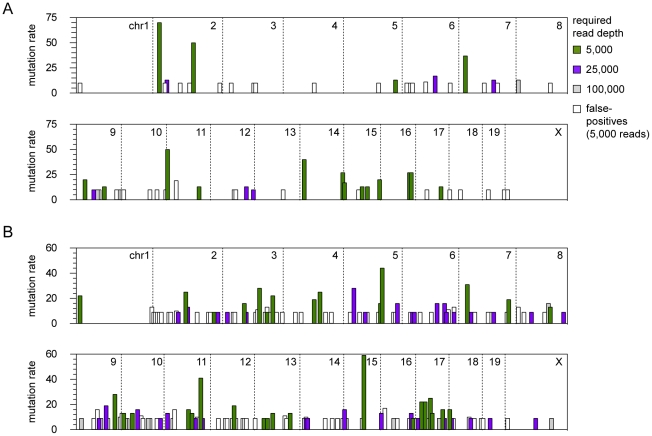
The recent development of the Sleeping Beauty (SB) system has led to the development of novel mouse models of cancer. Unlike spontaneous models, SB causes cancer through the action of mutagenic transposons that are mobilized in the genomes of somatic cells to induce mutations in cancer genes. While previous methods have successfully identified many transposon-tagged mutations in SB-induced tumors, limitations in DNA sequencing technology have prevented a comprehensive analysis of large tumor cohorts. Here we describe a novel method for producing genetic profiles of SB-induced tumors using Illumina sequencing. This method has dramatically increased the number of transposon-induced mutations identified in each tumor sample to reveal a level of genetic complexity much greater than previously appreciated. In addition, Illumina sequencing has allowed us to more precisely determine the depth of sequencing required to obtain a reproducible signature of transposon-induced mutations within tumor samples. The use of Illumina sequencing to characterize SB-induced tumors should significantly reduce sampling error that undoubtedly occurs using previous sequencing methods. As a consequence, the improved accuracy and precision provided by this method will allow candidate cancer genes to be identified with greater confidence. Overall, this method will facilitate ongoing efforts to decipher the genetic complexity of the human cancer genome by providing more accurate comparative information from Sleeping Beauty models of cancer.
Conflict of interest statement
Figures
Similar articles
-
Sequencing methods and datasets to improve functional interpretation of sleeping beauty mutagenesis screens.BMC Genomics. 2014 Dec 19;15(1):1150. doi: 10.1186/1471-2164-15-1150. BMC Genomics. 2014. PMID: 25526783 Free PMC article.
-
SB Digestor: a tailored driver gene identification tool for dissecting heterogeneous Sleeping Beauty transposon-induced tumors.Int J Biol Sci. 2023 Mar 13;19(6):1764-1777. doi: 10.7150/ijbs.81317. eCollection 2023. Int J Biol Sci. 2023. PMID: 37063417 Free PMC article.
-
Transposon-based screens for cancer gene discovery in mouse models.Semin Cancer Biol. 2010 Aug;20(4):261-8. doi: 10.1016/j.semcancer.2010.05.003. Epub 2010 May 15. Semin Cancer Biol. 2010. PMID: 20478384 Free PMC article. Review.
-
Identification of Cancer Genes Based on De Novo Transposon Insertion Site Analysis Using RNA and DNA Sequencing.Methods Mol Biol. 2019;1907:73-79. doi: 10.1007/978-1-4939-8967-6_5. Methods Mol Biol. 2019. PMID: 30542991
-
Sleeping Beauty transposon insertional mutagenesis based mouse models for cancer gene discovery.Curr Opin Genet Dev. 2015 Feb;30:66-72. doi: 10.1016/j.gde.2015.04.007. Epub 2015 Jun 4. Curr Opin Genet Dev. 2015. PMID: 26051241 Free PMC article. Review.
Cited by
-
Analyzing tumor heterogeneity and driver genes in single myeloid leukemia cells with SBCapSeq.Nat Biotechnol. 2016 Sep;34(9):962-72. doi: 10.1038/nbt.3637. Epub 2016 Aug 1. Nat Biotechnol. 2016. PMID: 27479497 Free PMC article.
-
Resistance mechanisms to TP53-MDM2 inhibition identified by in vivo piggyBac transposon mutagenesis screen in an Arf-/- mouse model.Proc Natl Acad Sci U S A. 2017 Mar 21;114(12):3151-3156. doi: 10.1073/pnas.1620262114. Epub 2017 Mar 6. Proc Natl Acad Sci U S A. 2017. PMID: 28265066 Free PMC article.
-
Divergent clonal selection dominates medulloblastoma at recurrence.Nature. 2016 Jan 21;529(7586):351-7. doi: 10.1038/nature16478. Epub 2016 Jan 13. Nature. 2016. PMID: 26760213 Free PMC article.
-
A Sleeping Beauty forward genetic screen identifies new genes and pathways driving osteosarcoma development and metastasis.Nat Genet. 2015 Jun;47(6):615-24. doi: 10.1038/ng.3293. Epub 2015 May 11. Nat Genet. 2015. PMID: 25961939 Free PMC article.
-
A Sleeping Beauty screen reveals NF-kB activation in CLL mouse model.Blood. 2013 May 23;121(21):4355-8. doi: 10.1182/blood-2013-02-486035. Epub 2013 Apr 16. Blood. 2013. PMID: 23591791 Free PMC article.
References
-
- Kool J, Berns A. High-throughput insertional mutagenesis screens in mice to identify oncogenic networks. Nat Rev Cancer. 2009;9:389–399. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
